Х-свързана лимфопролиферативни синдром момчета. болест на Дънкан
Х-свързана лимфопролиферативно синдром, наричан още болест на Дънкан (От името на семейството, в която за първи път е идентифициран) тя е Х-свързан рецесивен белег характеризира с увреден имунен отговор към Епщайн-Бар вирус.
Генетика и патогенеза на синдром X-свързана лимфопролиферативно - заболявания Дънкан. Дефектен ген е локализиран на дългото рамо на X хромозомата (сайт Xq23) и се клонира. Първоначално му протеинов продукт е наречен SAP (SLAM свързан протеин), но по-късно получи официално име SH2D1A. SLAM (сигнализация лимфоцитното активиране молекула - сигнализация лимфоцитното активиране молекула) е молекула адхезия.
Инфекциозни агенти и други стимуланти ускоряване неговия синтез в Т- и В-лимфоцити. SH2D1A е силно експресиран на тимоцити и T (за предпочитане на Th1) и NK-клетки от периферна кръв. Не е ясно дали това е в В-лимфоцити. По този начин, въпреки че заболяването често се наблюдава недостатъчност Duncan антитела всъщност засяга основната дефект на Т- и NK-клетки. SH2D1A конкурира с SHP-2 за свързване към SLAM протеин и по този начин действа като регулаторна молекула.
Липса на SH2D1A Той определя неконтролирана реакция цитотоксичен Т-лимфоцитен вирус инфекция на Epstein-Barr вирус. От SH2D1A протеин зависи от експресията на 2Ь4 молекули върху NK-клетки. Нарушаването на активирането на NK-клетки, медиирани от молекулата, също играе роля в устойчивостта нарушения по време на болест Дънкан.

Клиничните прояви на синдром X-свързана лимфопролиферативно - заболявания Дънкан
Х-свързана лимфопролиферативно синдром очевидно в момчетата само след инфекция с Епщайн-Бар вирус, обикновено до 5-годишна възраст. Има три основни клиничен фенотип: 1) светкавица, често фатално инфекциозна мононуклеоза (50%), 2) лимфом предпочитане В-клетки (25%) и 3) придобита хипогамаглобулинемия (25%).
продукти антитяло към антигена на ядрото на Епщайн-Бар рязко намалява, докато антитела към капсид антиген на вируса може да са дори много по-високи от нормалното. Прогнозата е неблагоприятно: 70% от пациентите умират от момчета под 10-годишна възраст. Известно е само 2 случаи, когато пациентите са живели повече от 40 години. При липса на семейна история на заболяването в диагностициран до усложнения е трудно да се установи, защото пациентите не правят никакви оплаквания. В същите семейства, засегнати болестта момчета могат да бъдат диагностицирани чрез ДНК анализ, преди първичния вируса.
Приблизително 50% от ограничен брой пациенти, трансплантация на HLA-идентичен нефракциониран костен мозък, сега продължават да живеят и признаци на заболяването те не притежават.
Има 2 семейства, всяка от които момчета един от последователите бе разкрито OVGGG, а от друга - фулминантен инфекциозна мононуклеоза. Въпреки това, всички пациенти във всяка от линиите на приемственост, въпреки различната клинична фенотип, имаше една и съща SH2D1A мутация. Следователно, всяко момче с диагноза OVGGG, особено когато има семейство от няколко пациенти от мъжки пол, трябва да се подозира синдром Х-свързан Лимфопролиферативната.
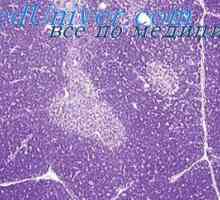 Комбиниран имунодефицит с fermentopathy. синдром nezelofa или alymphocytosis
Комбиниран имунодефицит с fermentopathy. синдром nezelofa или alymphocytosis Хипотеза два сигнала. Схема взаимодействие на Т и В лимфоцити
Хипотеза два сигнала. Схема взаимодействие на Т и В лимфоцити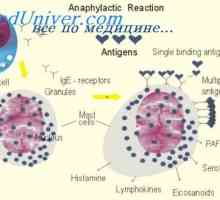 Индукционна IgE. Участие в IgE алергични реакции
Индукционна IgE. Участие в IgE алергични реакции Регулаторна функция на вродения имунитет. Контрол върху формирането на адаптивния имунитет
Регулаторна функция на вродения имунитет. Контрол върху формирането на адаптивния имунитет Х-свързан синдром хиперпродукция на имуноглобулин М (IgM) момчета. Мутация на CD40 CD154
Х-свързан синдром хиперпродукция на имуноглобулин М (IgM) момчета. Мутация на CD40 CD154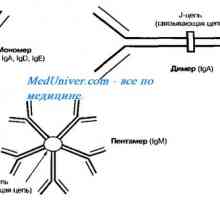 Автозомно рецесивен синдром хиперпродукция на имуноглобулин М (IgM). генна мутация помощ
Автозомно рецесивен синдром хиперпродукция на имуноглобулин М (IgM). генна мутация помощ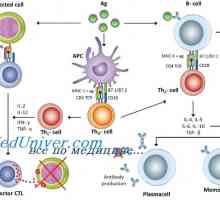 Механизми първичен имунен отговор в плода
Механизми първичен имунен отговор в плода Нарушенията на Т-лимфоцити. CD8 лимфопения
Нарушенията на Т-лимфоцити. CD8 лимфопения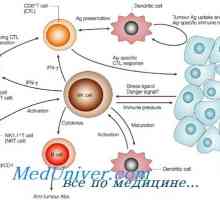 Образуване на NK-фетален имунни клетки. Т-лимфоцитна функция имунитет
Образуване на NK-фетален имунни клетки. Т-лимфоцитна функция имунитет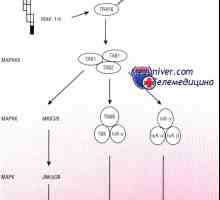 Схема IRF път в отговор на бактериите на чревния епител
Схема IRF път в отговор на бактериите на чревния епител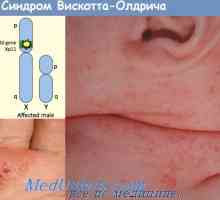 Синдром на Wiskott-Aldrich. Имунодефицитен с тромбоцитопения и екзема
Синдром на Wiskott-Aldrich. Имунодефицитен с тромбоцитопения и екзема Синдром на Омен. Immmunodefitsit при дефицит МНС антигени
Синдром на Омен. Immmunodefitsit при дефицит МНС антигени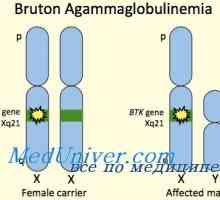 Х-свързана агамаглобулинемия. Brutonovskaya агамаглобулинемия деца
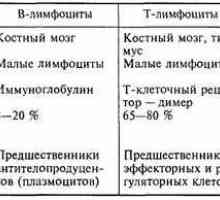
Х-свързана агамаглобулинемия. Brutonovskaya агамаглобулинемия деца Т-лимфоцити. Характеристики на Т-лимфоцити. Видове молекули по повърхността на Т-лимфоцити.
Т-лимфоцити. Характеристики на Т-лимфоцити. Видове молекули по повърхността на Т-лимфоцити.- Функция в клетки. Видове молекули върху повърхността на лимфоцитите.
 Произход (образуване) на клетките на имунната система. Функциите на клетките на имунната система.…
Произход (образуване) на клетките на имунната система. Функциите на клетките на имунната система.…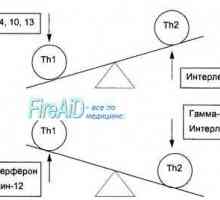 Т-лимфоцитна популация. Субпопулациите на Т-лимфоцити. CD4 Т-лимфоцити. CD8 Т-лимфоцити.
Т-лимфоцитна популация. Субпопулациите на Т-лимфоцити. CD4 Т-лимфоцити. CD8 Т-лимфоцити.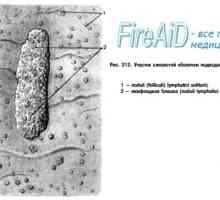 Лигавиците свързана лимфоидна тъкан. Лимфоидна тъкан на лигавицата.
Лигавиците свързана лимфоидна тъкан. Лимфоидна тъкан на лигавицата.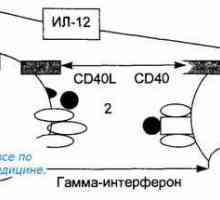 Антиген презентация. антиген разпознаване. Взаимодействие на Т-хелперните (Th1) с антиген…
Антиген презентация. антиген разпознаване. Взаимодействие на Т-хелперните (Th1) с антиген…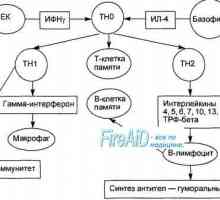 Активирането на Т и В лимфоцити в имунния отговор. Активирането на лимфоцити. Образува специфичен…
Активирането на Т и В лимфоцити в имунния отговор. Активирането на лимфоцити. Образува специфичен…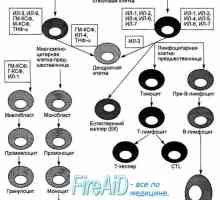 Образува специфичен имунен отговор. Имунологична памет като един вид имунен отговор.
Образува специфичен имунен отговор. Имунологична памет като един вид имунен отговор.