Въглехидратен метаболизъм при кърмачета

Съдържание
- Видео: Нарушаване на въглехидратния метаболизъм при деца
- заболявания съхранение Гликоген
- Видео: Филмът на манастира. Простите въглехидрати. Висок гликемичен индекс
- Нарушения на метаболизма фруктоза
- Нарушения на пируват метаболизъм
- Видео: патофизиология на въглехидратния метаболизъм (Лекция). Т. n. alhendi. част 2/2
- Видео: вродени нарушения на въглехидратния метаболизъм. Клинична картина, диагноза, лечение
- Други разстройства на въглехидратния метаболизъм
Галактоземията - автозомно-рецесивно заболяване се среща в 1 дете до 60 000 живородени деца.
В класическия, по-често под формата на дефицит на галактоза-1-fosfaturidiltransferazy, което води до натрупване на галактоза, галактоза-1-фосфат и galaktiola тъкан. По-рядко се дължи на дефицит галактоземия галактокиназен или епимеразната а. На допускане до лактоза настъпи при новородени храна повръщане, диария, хипербилирубинемия, хепатоспленомегалия, тубулна дисфункция, чернодробна недостатъчност, катаракта. Галактоза в кръвта на болни деца.
Диагнозата се потвърждава чрез количествено определяне на активността на галактоза-1-fosfatgransferazy в червените кръвни клетки, който ще бъде вярно, ако не се предхожда от червените кръвни клетки трансфузия.
Морфологични промени включват изразени с прогресивна чернодробна стеатоза psevdoatsinarnoy прегрупиране структура лобули пролиферация на жлъчните пътища, холестаза, фокална некроза с изход на цироза.
Освен това, има островни клетки хиперплазия, вакуолизация nephrothelial неспецифично исхемично увреждане на мозъка под формата на невронална смърт, глиоза, оток.
Морфологични промени на наследствена непоносимост към фруктоза и тирозинемия са сходни (вж. По-долу).
Нарушенията на фруктоза метаболизъм. Представлява заболяване с автозомно-рецесивно режим на наследяване, явен в новородено период за приемане с храна. Проявява се с повръщане, гадене, хепатомегалия, чернодробна недостатъчност, хипогликемия, лактатна ацидоза. Стеатоза, холестаза, портал фиброза и пролиферация cholangioles с трансформация на цироза.
Glycogenoses - група наследствени заболявания на въглехидратния метаболизъм в резултат от мутации в няколко гени, кодиращи ензими, които регулират синтеза и разграждането на гликоген в прости захари и нормална или анормална натрупването на гликоген в клетките на много тъкани. Класификацията се основава на дефект на определен ензим. Унаследява по автозомно рецесивен начин, с изключение гликогеноза тип VIII, секс-свързани.
Разнообразието от форми на болестта на съхранение гликоген (тип 14) отразява част в гликоген метаболизма на голям брой гени.
По-голямата част от пациентите имат системни прояви, наблюдавани при някои форми на неизпълнение на отделните органи. Характеризира се с симптоми на миопатия, както и пристъпи на хипогликемия, кардиомегалия развитие.
Видео: Нарушаване на въглехидратния метаболизъм при деца
заболявания съхранение Гликоген
заболявания съхранение гликоген (GSDs) поради недостатъчност на ензими, участващи в синтеза или унищожаване glikogena- недостатъци могат да се появят в черния дроб, мускулите и предизвикват хипогликемия или отлагане на необичайни количества или видове анормален гликоген в тъканите.
Видео: Филмът на манастира. Простите въглехидрати. Висок гликемичен индекс
Наследяване на GSDs автозомно рецесивен освен видове GSD VIII / IX са Х-свързана. Честотата се изчислява на около 1/25 000 раждания, а тя може да се подценява, тъй като светлинните субклинични форми не винаги са диагностицирани.
Възраст на настъпване, клинични прояви и тежест зависи от вида.
Диагнозата се потвърждава със значително намаление на ензимната активност в черния дроб (типове I, III, VI и VIII / IX), мускулите (тип Пб, III, VII и VIII / IX), кожни фибробласти (Видове Ха и IV) или еритроцити (тип VII) отсъствие или увеличаване на активността на венозна лактат / исхемия ръка (тип V и VII). Прогноза и лечение варира в зависимост от вида, но лечението често включва хранителни добавки с царевично нишесте за да се осигури постоянен източник на глюкоза в черния дроб форми на GSD и упражнения за предотвратяване на мускулни форми.
Дефекти в гликолиза (рядко) може да предизвика синдром подобен на GSDs. Фосфоглицерат киназа дефицит, фосфоглицерат мутаза и лактат миопатия тип имитират GSD V и VII- дефицити глюкозен транспорт протеин 2 (сайдер Fanconi - Bickel) имитират GSD хепатопатия други видове (например, 1111, IV, VI).
Нарушения на метаболизма фруктоза
Недостигът метаболизиращ ензим фруктоза, може да бъде асимптоматични или да причини хипогликемия.
Фруктоза е монозахарид настоящото във високи концентрации в плодове и мед и съставлява част от захароза и сорбитол.
Недостигът на фруктоза-1-фосфат алдолаза (алдолаза В). Този дефицит е клиничен синдром, наследствена непоносимост към фруктоза. Наследяването е автозомно retsessivnoe- честота се оценява на 1/20 000 раждания. Бебетата са в добро здраве, докато не се консумира фруктоза. Продължителната употреба може да доведе до цироза, умствено влошаване и тубулна ацидоза проксималния липсата на фосфат в урината и глюкоза.
Диагностика на базата на симптоми предполагат, с оглед на последните прием на фруктоза и потвърди ензимен анализ на чернодробната тъкан биопсия или чрез индуциране на хипогликемия интравенозна инфузия на фруктоза в количество от 200 мг / кг.
Краткосрочни лечението е да се получи глюкоза gipoglikemii- дългосрочно лечение е премахването на консумацията на фруктоза, захароза и сорбитол. Много от пациентите развиват естествена непоносимост към фруктоза храна. При лечението прогнозата е отлична.
фруктокиназа дефицит. Този дефицит води до доброкачествена увеличение на кръв и урина нива на фруктоза (доброкачествена fructosuria). Наследяването е автозомно retsessivnoe- честота е около 1/130 000 раждания.
Статус безсимптомно и диагностицира случайно при откриването на пикочните neglyukozovosstanavlivayuschego вещества.
Недостигът на фруктоза-1, 6-бисфосфатаза. Този дефицит компрометира глюконеогенезата и води до бърза хипогликемия, кетоза и ацидоза. Недостигът може да бъде фатално за бебета. Наследяването автозомно retsessivnoe- честота е неизвестна. Има епизоди на треска.
Спешно лечение се състои от перорално или интравенозно приложение на глюкоза. Толерантност към глад обикновено се увеличава с възрастта.
Нарушения на пируват метаболизъм
Невъзможност да метаболизира пируват води до развитието на лактатна ацидоза и различни нарушения на централната нервна система.
Видео: патофизиология на въглехидратния метаболизъм (Лекция). Т. N. Alhendi. част 2/2
Пируват е важен субстрат на въглехидратния метаболизъм.
Недостигът на пируват дехидрогеназа. Piru vatdegidrogenaza е мултиен комплекс, отговорен за образуването на ацетил-СоА от пируват за цикъла на Кребс. Недостигът води до повишени нива на пируват и следователно повишаване на нивото на млечна киселина. Х-свързан наследство или авто-хромозомна рецесивна.
Видео: вродени нарушения на въглехидратния метаболизъм. Клинична картина, диагноза, лечение
Клиничните прояви варират по тежест, но включват лактатна ацидоза.
Диагнозата се потвърждава чрез ензимен анализ активност кожата фибробластен ДНК анализ на тези два метода.
Ясно е, че не съществува ефективно лечение, а на диета с ниско съдържание на въглехидрати или кетогенна диета и хранителни добавки тиамин са полезни за някои пациенти.
Недостигът на пируват карбоксилаза. Недостигът може да бъде първична или вторична недостатъчност golokarboksilazy синтетаза биотин или biotinidazy- автозомно рецесивно унаследяване на, и двата варианта доведат до лактатна ацидоза.
Честотата на първичния дефицит <1/250 000 родов, но может быть выше у некоторых американских индейских народов. Задержка психомоторного развития с эпилептическими припадками и спастичность являются основными клиническими проявлениями. Нарушения лабораторных показателей включают гипераммониемию- молочный ацидоз, кетоацидоз.
Клиничните прояви на вторична недостатъчност са подобни и включват неуспех да се развиват, припадъци, и други органични aciduria.
Ефективното лечение не съществува, но някои пациенти с първични дефицити и всички лица, които имат среден тежестта на заболяването, е необходимо да се добави биотин 5-20 мг орално веднъж дневно.
Други разстройства на въглехидратния метаболизъм
Недостигът на фосфоенолпируватни карбоксиназа (261,680) дава глюконеогенезата и води до симптоми и признаци, подобни на чернодробна форма гликогеноза, но без натрупването на гликоген в черния дроб.
Други дефицити включват недостатъчност гликолитични ензими или ензими на пентозофосфатния път. Типични примери са дефицит пируват киназа и глюкозо-6-фосфат дехидрогеназа (G6PD), което може да доведе до развитието на хемолитична анемия. Wernicke синдром - Korsakoff причинени частично недостатъчност транскетолаза пентозофосфатния път е ензим, изисква тиамин и като кофактор.
 Laktofiltrum диария
Laktofiltrum диария Poslezheltushnaya енцефалопатия. Microspherocytosis наследствено заболяване Минковски-Chauffard
Poslezheltushnaya енцефалопатия. Microspherocytosis наследствено заболяване Минковски-Chauffard Галактоземия
Галактоземия Гликоген болестни съхранение морбили, Андерсен McArdl. болест Хърси е, Thomson, контейнери
Гликоген болестни съхранение морбили, Андерсен McArdl. болест Хърси е, Thomson, контейнери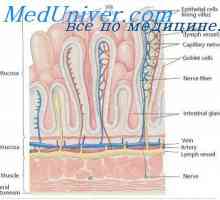 Въглехидратната абсорбция в червата. Усвояването на протеините в червата
Въглехидратната абсорбция в червата. Усвояването на протеините в червата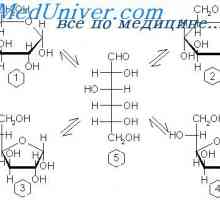 Физиологията на глюкозния метаболизъм. Транспорт на глюкоза в клетъчната мембрана
Физиологията на глюкозния метаболизъм. Транспорт на глюкоза в клетъчната мембрана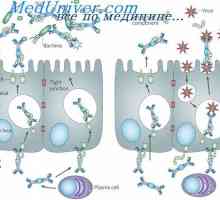 Секрецията на имуноглобулини. Етапи секреция антитяло
Секрецията на имуноглобулини. Етапи секреция антитяло Автозомно-рецесивно поликистоза на бъбреците при деца. Диагностика и лечение
Автозомно-рецесивно поликистоза на бъбреците при деца. Диагностика и лечение Неонатална жълтеница с метаболитни нарушения
Неонатална жълтеница с метаболитни нарушения Методи за оценка на метаболизма на въглехидрати и мазнини в тялото
Методи за оценка на метаболизма на въглехидрати и мазнини в тялото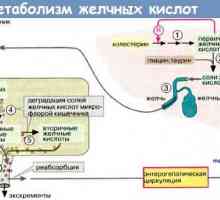 Прогресивно фамилна чернодробна холестаза (PFIC) видове, диагностика
Прогресивно фамилна чернодробна холестаза (PFIC) видове, диагностика Лактулозата (lastulosum). Лактулозата отнася до синтетичен дизахарид. Когато се прилага орално не…
Лактулозата (lastulosum). Лактулозата отнася до синтетичен дизахарид. Когато се прилага орално не…- Ehovist (echovist). Ehovist-200 е суспензия на микронизиран г-galaktoy. Когато суспендира…
- Здраве Енциклопедия, болест, лекарства, лекар, аптека, инфекция, резюмета, пол, гинекология,…
- Терапия, заболяване на храносмилателната система
 Нарушения на метаболизма на аминокиселина
Нарушения на метаболизма на аминокиселина Атрезия на жлъчните пътища
Атрезия на жлъчните пътища Обмен нарушения метали
Обмен нарушения метали Неонатален холестаза при деца: причините, лечение, симптоми
Неонатален холестаза при деца: причините, лечение, симптоми Метаболитни нарушения в метаболизма на пикочната киселина и метали
Метаболитни нарушения в метаболизма на пикочната киселина и метали Цироза на черния дроб в детските симптоми
Цироза на черния дроб в детските симптоми