Мозъчния рак на щитовидната жлеза

Съдържание
Видео: мозъчния и добре диференциран рак на щитовидната жлеза: иновативни прогностични, предсказващи и терапевтични подходи
На MTC съставлява 5-10% от всички злокачествени тумори на щитовидната жлеза.
Приблизително 75% от МТС е спорадично, а останалите - семейството, свързани с един от трите генетични синдроми: семейство изолира МТС, множествена ендокринна неоплазия ON [MEN PA (МТС, феохромоцитом и първичен хиперпаратироидизъм)] или MAN PB [МТС, феохромоцитом , множествена лигавицата невроми и (рядко) първичен хиперпаратироидизъм].
В сърцето на МТС онкогенни мутации са RET, разположен върху хромозома 10q11.2. RET ген кодира мембранен рецептор тирозин киназна активност, който лиганди принадлежат към семейството на невротрофични фактори, продуцирани от глиални клетки (GDNF). Този рецептор се експресира по време на развитието на мигриращите невралната тръба клетки, които пораждат невроендокринни клетки (В-клетки и клетките на надбъбречната медула) и клетки парасимпатиковата и симпатиковата ганглии на периферната нервна система. Различни мутации в RET са отговорни за пет различни синдроми. В сърцето на БКП MEN MTC и семейството са някои от активиращи мутации в този ген. Други активиращи мутации водят до MAN PB. Повече от половината от случаите на спорадична MTC настъпва клонална соматична мутация (т. Е. Само открива в туморни клетки), идентичен с една от мутациите причиняват фамилни форми на МТС. Gene прегрупиране RETc образуват химерен ген често се наблюдава в клетки на папиларен рак на щитовидната жлеза. Експерименти с трансгенни мишки показват, че преструктурирането на гена на RET е достатъчно за развитието на рак на папиларен тироиден. Накрая, инактивиращи мутации в заболяване RET причина на Hirschsprung - вродена липса на парасимпатиковата ганглии на корема, придружени от нарушение на чревната подвижност, което води до гигантизъм на дебелото черво (мегаколон).
Видео: Нова ера в терапията за напреднали медуларен карцином на щитовидната жлеза
Клиничните прояви на множество ендокринни синдроми neloplazii
MEN I
Хиперплазия на паращитовидните жлези (много често)
Панкреаса тумор (доброкачествен или злокачествен)
- Gistrinoma
- инсулином
- Глукагономи, VIP-om (и двете рядко)
тумори на хипофизата
- Секретиращи растежен хормон
- пролактин-секретиращи
- ACTH-секретиращ
Други тумори: липома, карциноид, надбъбречна аденом и щитовидната жлеза
MEN IIA
Мозъчния рак на щитовидната жлеза
Феохромоцитом (доброкачествени или злокачествени)
Хиперплазия на паращитовидните жлези
MEN IIB
Мозъчния рак на щитовидната жлеза
феохромоцитом
Неуроми мускулна ganglioneuroma
Marfanopodobnaya външен вид
Хиперпаратиреоидизмът (много рядко)
} {Модул direkt4
MTC обикновено се локализира в центъра или в горната част на лоба на щитовидната жлеза. В единични случаи, то обикновено се развива само в един от сегментите, но често многоцентрично и двустранни когато семейството форми. Хистологично MTC рано разграничи от други видове рак на щитовидната жлеза с наличието на амилоид (еозинофилен вещество, оцветени с Конго червено). След това се оказа, че амилоид се състои от гъсти фибриларни протеинови отлагания, които формират един сгънат слой (-структура). Когато този протеин е MTC прокалцитонин или самата калцитонин. Ето защо, морфологична диагностика на МТС, определен от имунохистохимично оцветяване на калцитонин.
MTC се проявява по различни начини. Спорадични тумори са едновременно много агресивен и изключително maloaktivnymi- 5-годишната преживяемост е 50%. По отношение на семейните форми, това зависи от тях за синдроми. Най-агресивен МТС се случва в този синдром MEN IIB- да оцелее не повече от 50% от пациентите в рамките на 2 години. В MEN IIA за MTC наподобява тази спорадични форми на рак. Най-малко агресивни потоци изолирани фамилна МТС. Туморът може да се разпространи и в регионалните лимфни възли или да се даде хематогенен метастази в белите дробове и други органи. Метастазни туморни понякога съпроводено от хронична диария, патогенеза от които е неясно. Освен калцитонин, тези тумори секретират различни други биологично активни вещества, включително простагландини, серотонин, хистамин и пептидни хормони (АСТН, соматостатин, кортикотропин-освобождаващ фактор). В някои случаи диария елиминира дългодействащ соматостатиновия аналог (октреотид), който блокира секрецията от него.
Калцитонин - MTC маркер. По-голяма надеждност на маркера получава в стимулиране на секрецията. Обикновено това се прави с помощта на пентагастрин (0.5 мкг / кг интравенозно 5 секунди) или бърза инфузия на калциев глюконат (2 мг / кг в продължение на 1 минута). Вземат се кръвни проби преди и след 1, 2 и 5 минути след стимулиране. За да се увеличи теста за чувствителност извършва комбиниран: калциев след инфузия незабавно прилага пентагастрин. В ранните етапи на тумор базово ниво калцитонин често е нормално, но при метастазирал MTC първоначалната концентрация на калцитонин е много пъти по-висока от нормалното. Въпреки това, съдържанието на калций в серума не се променя. Тумор обикновено отделя калцитонин форма високо молекулно тегло и по-малко биологична активност, но често увеличава ниво мономер и калцитонин. В повечето случаи, вместо анализ проба стимул извършва онкоген RET, но базално калцитонин поддържа неговата стойност като индикатор на туморна активност.
Всички членове на семейството на RET мутации пациент за подпомагане трябва да бъдат изследвани за МТС и връзката му с синдроми MEN IIA и IIB MEN. В идентифициране на мутации на този ген обикновено препоръчвам общо tireoidekto-мисията, преди развитието на рак или да повишат нивото на калцитонин. Децата в семейства с известна мутация в RET се препоръчва да разследва носителя на мутация веднага след раждането. При откриване на мутации се провежда общо тиреоидектомия с лимфна дисекция, а при липса на мутация допълнително проучване е необходимо. Въпреки това, времето за профилактично тиреоидектомия при асимптоматични носители на мутации RET зависи от точното генотип (т. Е. мутант кодон на гена) и клинични обстоятелства. В случаите на специалисти с висок риск препоръчваме да извърши операцията, докато 6-12 месечна възраст, със среден риск - до 5-годишна възраст и при нисък риск тиреоидектомия може да се забави до 10-годишна възраст. Като основа за повече от 95% от случаите на наследствен МТС и 25% от спорадични тумори е ограничен брой генни мутации в RET, могат да се извършват проучвания, използвайки стандартни комплекти.
Проучване на членовете на семейството трябва да бъде още по-очевидно с спорадично МТС, като почти 25% от новите случаи на пробанд принадлежат към семейства с превоз на наследствен синдром. Както и в известен семейство МТС в близки провокация тестове, извършени върху секрецията на калцитонин или анализ на ДНК в туморните клетки и други клетки за мутации на RET. Идентификация на мутации в туморната тъкан показва само своята соматични и тумор - спорадично. Наличието на същата мутация в тумора, и геномна ДНК показва фамилна форма на заболяването и изисква внимателно изследване на всички членове на семейството.
 Хепатоцелуларен карцином и рак на дебелото черво при деца. Тумори на щитовидната жлеза и…
Хепатоцелуларен карцином и рак на дебелото черво при деца. Тумори на щитовидната жлеза и… Функциите на хормони на щитовидната жлеза. Влияние на хормони на щитовидната жлеза на метаболизма
Функциите на хормони на щитовидната жлеза. Влияние на хормони на щитовидната жлеза на метаболизма Хормони на щитовидната жлеза. Синтезът на хормони на щитовидната жлеза, йод капан
Хормони на щитовидната жлеза. Синтезът на хормони на щитовидната жлеза, йод капан Злокачествените новообразувания на симптоми на щитовидната, класификация
Злокачествените новообразувания на симптоми на щитовидната, класификация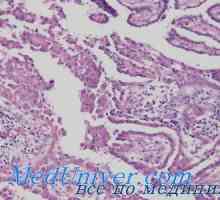 Гиосарком или хемангио на щитовидната жлеза. Neyrosarkoma или злокачествена шванома на щитовидната…
Гиосарком или хемангио на щитовидната жлеза. Neyrosarkoma или злокачествена шванома на щитовидната…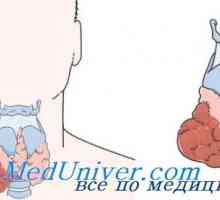 Плоскоклетъчен (neorogovevayuschy) рак на щитовидната жлеза. Nizkodifferentsirovaniaya форма на рак…
Плоскоклетъчен (neorogovevayuschy) рак на щитовидната жлеза. Nizkodifferentsirovaniaya форма на рак…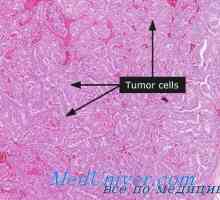 Clasmocytoma и лимфома на щитовидната жлеза. Флуоресцентна микроскопия на щитовидната жлеза
Clasmocytoma и лимфома на щитовидната жлеза. Флуоресцентна микроскопия на щитовидната жлеза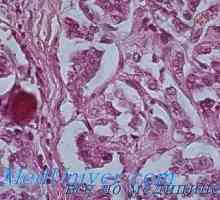 Hyperparathyroid генерализирана влакнест остеодистрофия (болест на фон Реклингхаузен) морфология,…
Hyperparathyroid генерализирана влакнест остеодистрофия (болест на фон Реклингхаузен) морфология,… Миграцията нарушения на ентерично нервна система при болест на Hirschsprung
Миграцията нарушения на ентерично нервна система при болест на Hirschsprung Учените са открили ген свързване на рак на гърдата и на щитовидната жлеза
Учените са открили ген свързване на рак на гърдата и на щитовидната жлеза- В Европа одобри ново лекарство за лечение на рак на щитовидната жлеза lenvima
- Ефект на мутации в задържане на ефективността на терапията медуларен рак на щитовидната жлеза
- Ендокринна система: ключови думи
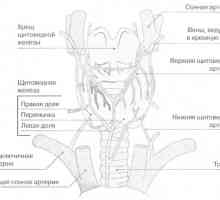 Щитовидната жлеза и паращитовидните жлези
Щитовидната жлеза и паращитовидните жлези- Синдромът на множествена ендокринна неоплазия тип 1
- Общи характеристики на синдроми на автоимунни полижлези
- Тумори. Ендокринната туморни заболявания характер см. Акромегалия, virilnoe синдром,…
- Здраве Енциклопедия, болест, лекарства, лекар, аптека, инфекция, резюмета, пол, гинекология,…
 Автоимунно заболяване на щитовидната жлеза
Автоимунно заболяване на щитовидната жлеза Безболезнено тиреоидит на рак на щитовидната жлеза: симптоми, лечение, причини
Безболезнено тиреоидит на рак на щитовидната жлеза: симптоми, лечение, причини Синдром на множествена ендокринна неоплазия, Тип II (Мейн 2): причини, симптоми, лечение, симптоми
Синдром на множествена ендокринна неоплазия, Тип II (Мейн 2): причини, симптоми, лечение, симптоми