Gangliosidosis. Sandhofa заболяване и ювенилен gangliosidosis
при gangliosidosis натрупване в тъканите на глюкозилцерамид главно кисела - киселина ганглиозид.
За да се разбере модерното класификация ганглиозид трябва да знаете, символите, които отразяват различията в тяхната биохимична структура. Буквата G посочва ганглиосид и буквите М, D и Т брой молекули на сиалова киселина.
Специално ултраструктурната проучване на невроните кора (Пирамидалния клетъчен) като neyrolipidozah показва присъствието на така наречените meganevritov --expanded придатъци, разположени между тялото и аксон на неврон. Вътрешни meganevritov шипове са образувани, които са частично в контакт един с друг, образувайки синаптичните везикули. Значителна честота на аберантни синапси наблюдавани главно в класическия вариант Tay - Sachs заболяване (GM2) и субстрата е морфологични лезии невронна функция [пурпура D., Sazuki K, 1976]. Хистохимична проучване не даде точно описание на характера на липиди. Включване с GM2 PAS-позитивни, да се получи червено оцветяване с крезилвиолет, слабо положителен с Судан черно.
патологична диагноза може да се направи въз основа на ректални биопсия - промени в балон субмукозно неврони (meyssnerovo) плексус. Възможно е да се установи диагнозата в prenatalyyum период на базата на амниотични клетки за анализ на данни и околоплодна течност, която обикновено по време на второто тримесечие на бременността трябва да съдържат хексозаминидаза А и В, Тей -Saksa фракция А отсъства.

Sandhofa заболяване - Тип 2-GM2 gangliosidosis, автозомно рецесивно наследствено заболяване, причинено от пълен набор от части А и хексозаминидаза. В допълнение към ганглиозид GM2, малко количество от натрупаните други гликолипиди. В клиничното протичане и симптомите не се различават от Tay - Sachs заболяване. Морфологични разграничение е да се открие не само натрупване на клетки в нервната система, но няколко макрофаги и висцерални органи, както и в nephrothelial.
ювенилен gangliosidosis, Тип 3-GM2 gangliosidosis, развива поради непълно блок фракция А хексозаминидаза. Стартиране на възраст от 2-6 години и завършва със смърт на възраст от 5-15 години. Останалата част от промените са същите, както в детска формата на Tay-Sachs заболяване. Освен тези три форми GM2 gangliosidosis с установена ензим дефект, има описания на много случаи, подобни на amavroticheskoi идиот но различаващ се от него, както в редица клинични прояви и продължителността на заболяването и от топографията и ултраструктурата на липидни отлагания, както и биохимичен състав аз спаси катаболните продукти.
В момента може да се покаже, че някои форми amavroticheskoi инфантилен идиотизъм (Hegbsrga тип - Santavuori), късна инфантилен (тип Yanovsky - Bilshovskogo), ювенилен (тип Shpilmeera - Vogt) и възрастни (тип Kufs) принадлежат към т.нар negangliozidoznoy amavroticheskoi идиотизма, тъй като тези четири форми в цитоплазмата на клетки от различни тъкани разкрити включване състояща се от ceroid липофуксин lipopigmentov, фината структура на която е различна, обаче, от липофусциновите пигменти и ceroid инволютивна характер. В основата на всичките четири форми на заболявания все още не е ясно, ензимен дефицит. В допълнение, има все още са така наречените нетипични и междинни форми.
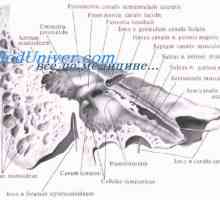 Вегетативната нервна система на ембриона. На парасимпатиковата нервна система на плода
Вегетативната нервна система на ембриона. На парасимпатиковата нервна система на плода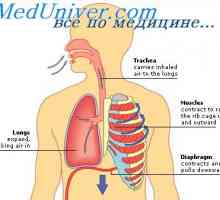 Mucolipidosis: mannozidoz и fucosidosis. Младежката sulfatidoz тип oustina
Mucolipidosis: mannozidoz и fucosidosis. Младежката sulfatidoz тип oustina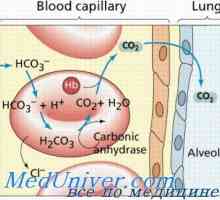 Amavroticheskaya вродена идиотия. Mucolipidosis
Amavroticheskaya вродена идиотия. Mucolipidosis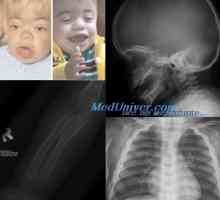 Класификация на мукополизахаридоза. На Hurler заболяване, gentera, Sanfilippo, Morquio
Класификация на мукополизахаридоза. На Hurler заболяване, gentera, Sanfilippo, Morquio Гликоген болестни съхранение морбили, Андерсен McArdl. болест Хърси е, Thomson, контейнери
Гликоген болестни съхранение морбили, Андерсен McArdl. болест Хърси е, Thomson, контейнери Резултати в активирането на лимфоцитите. Промени в клетките след активиране
Резултати в активирането на лимфоцитите. Промени в клетките след активиране Хипербарна кислород. Организация на нервната система
Хипербарна кислород. Организация на нервната система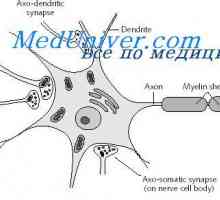 Ниско молекулно скорост медиатори. ацетилхолин
Ниско молекулно скорост медиатори. ацетилхолин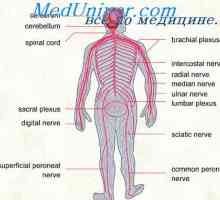 Подкорова ниво на нервната система. Кортикалното ниво на нервната система
Подкорова ниво на нервната система. Кортикалното ниво на нервната система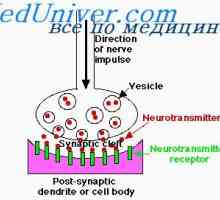 Неврони облекчение. функция на дендрити
Неврони облекчение. функция на дендрити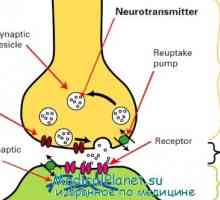 Физиология на нервните синапси. анатомия на синапс
Физиология на нервните синапси. анатомия на синапс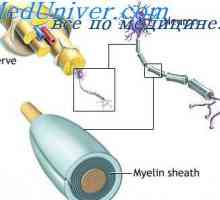 Механизмите на предаването на нервните сигнали. Прагови и подпрагови нервните стимули
Механизмите на предаването на нервните сигнали. Прагови и подпрагови нервните стимули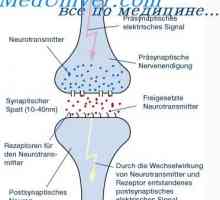 Най-възбудено състояние на неврона. Умората на синоптични
Най-възбудено състояние на неврона. Умората на синоптични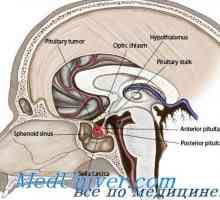 Фоликулостимулиращ хормон FSH
Фоликулостимулиращ хормон FSH Невроните: ключови термини
Невроните: ключови термини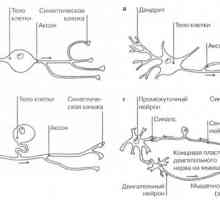 Видове нервните клетки
Видове нервните клетки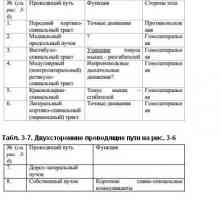 Гръбначния Анатомия
Гръбначния Анатомия- Серотонин или 5-хидрокситриптамин, в химическата структура на групата indolylalkylamines. Той е…
 Нарушение на липидния метаболизъм
Нарушение на липидния метаболизъм Неврофизиологични механизми за възбуждане на визуалното пътека
Неврофизиологични механизми за възбуждане на визуалното пътека Neyrometabolicheskie заболяване
Neyrometabolicheskie заболяване