Mucolipidosis: mannozidoz и fucosidosis. Младежката sulfatidoz тип oustina
с изключение на gangliosidosis GM1, Допълнителни изследвания ще подчертаят три нозологични форми Mucolipidosis с установените ензимни дефекти - mannozidoz, fucosidosis sulfatidoz и непълнолетни (тип Oustina).
Mannozidoz - ензим дефект е ограничен активност на а-манозидаза, което пречи на катаболизма на маноза-богата част glikoprotsidov и клетки (главно на черния дроб и мозък) се натрупва олигозахарид богата на маноза. Заболяването е описано в деца 2-4.5 години проявява психомоторно забавяне на развитието, хепатоспленомегалия, kyphoscoliosis, помътняване на лещата, вакуолизация в лимфоцити от периферна кръв. Смъртта настъпва в предучилищна възраст години.
аутопсия проучване невронални определени промени, подобни на промените в amavroticheskoy идиотия локализирани на багажника и в кората на главния мозък, гръбначния мозък, с явленията на демиелинизация и глиоза следват. Промени в ретината на окото отсъстват. натрупване на клетки са открити, освен това, в черния дроб, далака, костния мозък.
отлагам вещество частично разтворим във водни разтвори, PAS-позитивни дава отговор на липиди. Електрон-микроскопско изследване на лимфоцити и в невроните открива уголемени сферични лизозоми - вакуола натрупване.

fucosidosis - ензим дефект е напълно инактивира-фукозидоза и арилсулфатаза А [Troost J. и др, 1977] В мозъка, черния дроб и други органи, което води до натрупване на гликолипиди (tseramidoligosaharidov), съдържащи фукоза едновременно натрупват и киселинни мукополизахариди (хексуронова киселина). Наследяването е автозомно-рецесивно.
болест описан в SIBs на 2-3 години, характеризиращ се чрез увеличаване спастична парализа, умствена изостаналост, кахексия.
при fucosidosis наблюдавания кардиомегалия, атрофични изменения на кожата и скромни промени hondrodistroficheskogo тип. Намерени в урината повишено съдържание фукозид в периферните кръвни лимфоцити - вакуолизация. Има съобщения за две клинични видове fucosidosis в зависимост от степента на ензимната активност [Troost J. и сътр., 1977].
промени в мозъка напомня sudanofilnuyu левкодистрофия с едновременно загуба на неврони в мозъка и малкия мозък. Lipidnos вещество се натрупва около кръвоносните съдове. натрупване на клетки са открити, освен това, в черния дроб, миокарда, бъбреците, белите дробове, кожата, конюнктивата на окото. Electron микроскопични промени в натрупването на клетки наподобяват синдром на Hurler (големи вакуолен лизозоми) и липидоза (наличие и концентрични ламеларни структури). Хистохимично включвания съдържат олигозахаридни вериги.
може да открива дефицит на L-фукозидаза в една култура на кожни фибробласти в клетките на амниона и околоплодните води, което дава възможност да се определи пренатална диагностика fucosidosis.
юношески sulfatidoz (Тип Oustina) е комбинация от промени характеристика метахроматична левкодистрофия (sulfatidoza) и gargoilizma (мукополизахаридоза). Болест harakteruzetsya дефицит на активност на ензим арилсулфатаза А, В и С, за разлика от класическата метахроматична левкодистрофия, което пречи на активността на само арилсулфатаза А. При деца, с изключение на тези, специфични за метахроматична левкодистрофия, има промени в лицевите кости, гърдите и дългите кости, типичен за синдром на Hurler. Урината разпределени дерматан сулфат, хепарансулфатът и сулфатиди. Тези вещества се отлагат в тъканите.
В допълнение към тези форми на Mucolipidosis на определени ензимни дефекти описани изпълнения tezaurismozov смесен тип, които се различават едни от други клинични и морфологични характеристики доведе до различна степен на увреждане на скелета и вътрешностите и различна степен на увреждания на централната нервна система. Автозоми наследил рецесивен. По-подробни три възможности: тип I, тип II, тип III (psevdopolidistrofiya).
 Poslezheltushnaya енцефалопатия. Microspherocytosis наследствено заболяване Минковски-Chauffard
Poslezheltushnaya енцефалопатия. Microspherocytosis наследствено заболяване Минковски-Chauffard Pathways ембрион. Образуването на проводима пътеки плода
Pathways ембрион. Образуването на проводима пътеки плода Gangliosidosis. Sandhofa заболяване и ювенилен gangliosidosis
Gangliosidosis. Sandhofa заболяване и ювенилен gangliosidosis Болест Galaktoziltseramidoz Крабе. Sulfatidoz метахроматна leykodistrosriya Scholz
Болест Galaktoziltseramidoz Крабе. Sulfatidoz метахроматна leykodistrosriya Scholz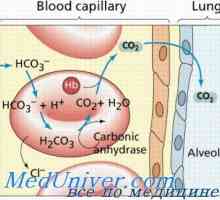 Amavroticheskaya вродена идиотия. Mucolipidosis
Amavroticheskaya вродена идиотия. Mucolipidosis Болест Lipogranulematoz Farber. Диагностика на Farber заболяване
Болест Lipogranulematoz Farber. Диагностика на Farber заболяване Uolmana генерализирана xanthelasmatosis заболяване. синдром refzuma
Uolmana генерализирана xanthelasmatosis заболяване. синдром refzuma И левкодистрофии мукополизахаридози. Опасност от Мукополизахаридоза
И левкодистрофии мукополизахаридози. Опасност от Мукополизахаридоза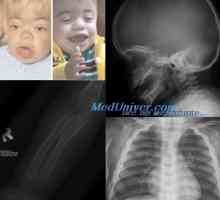 Класификация на мукополизахаридоза. На Hurler заболяване, gentera, Sanfilippo, Morquio
Класификация на мукополизахаридоза. На Hurler заболяване, gentera, Sanfilippo, Morquio Гликоген болестни съхранение морбили, Андерсен McArdl. болест Хърси е, Thomson, контейнери
Гликоген болестни съхранение морбили, Андерсен McArdl. болест Хърси е, Thomson, контейнери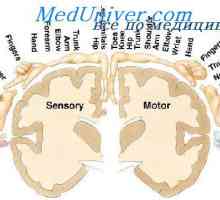 Прехвърляне на възбуждане от кората на главния мозък към мускулите. Кортикоспиналните (пирамидална)…
Прехвърляне на възбуждане от кората на главния мозък към мускулите. Кортикоспиналните (пирамидална)…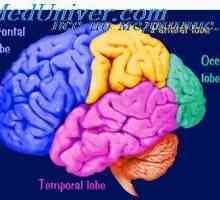 Кората на главния мозък. Физиологични анатомия на кората на главния мозък
Кората на главния мозък. Физиологични анатомия на кората на главния мозък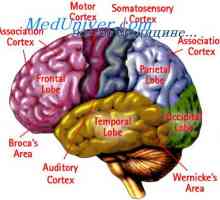 Съобщение на кората на главния мозък с други отдели. Специфични области на кората на главния мозък
Съобщение на кората на главния мозък с други отдели. Специфични области на кората на главния мозък Ретикуларна дисгенезис. пуриновите нуклеозиди дефицит фосфорилаза
Ретикуларна дисгенезис. пуриновите нуклеозиди дефицит фосфорилаза В-лимфоцити. Характеризиране на В-лимфоцити. памет клетки.
В-лимфоцити. Характеризиране на В-лимфоцити. памет клетки. Болест на Гоше се отнася до заболявания sfingolipidozam lipidov- натрупване поради дефект на гена,…
Болест на Гоше се отнася до заболявания sfingolipidozam lipidov- натрупване поради дефект на гена,…- Амблиопия истерия. Етиология. Инхибирането на визуалното възприятие в кората на главния мозък
 Нарушение на липидния метаболизъм
Нарушение на липидния метаболизъм Niemann-Pick заболяванията
Niemann-Pick заболяванията Neyrometabolicheskie заболяване
Neyrometabolicheskie заболяване Mucolipidosis
Mucolipidosis