Нематода-стоп-половата синдром. Генетика на мъжкото безплодие
Нематода-стоп-половата синдром - автозомно доминантно заболяване, характеризиращо се с I скъсяване на пръстите на ръцете и краката, пръстите клинодактилия V, непълна слят Мюлеров (по този начин евентуално удвояване на шийката на матката и вагинални стени, образуващи надлъжната) и аномалии в бъбреците. Мъжете могат да хипоспадия. Нематода-стоп-половата синдром се причинява от мутации NOHA13 ген и се наследява по автозомно доминантен начин. Той може да предизвиква мутация води до хаплоинсуфициенция NOHA13 ген.
NOHA13 - представителна група от НОХ транскрипционни фактори, изразена в дисталните части на крайниците и Мюлеров. Неговата роля в сливането на Мюлеров и тяхната диференциация не е напълно ясно. синдром Frinsa - симптом на няколко малформации, включително разделяне на горната устна и небцето, дефекти на ЦНС, малформация на пикочните пътища, сърцето и нарушаването на сливането Мюлеров. Обикновено това е подробна и се унаследява по автозомно рецесивен.
Най-добре познат до момента генетични синдроми, свързани с аномалии на репродуктивния тракт, причинени от недостиг на ключови ензими на надбъбречните жлези или тестисите, наследена в автозомно рецесивен начин, или дефект AR. Надбъбречна недостатъчност ензими, такива като 21-хидроксилаза води до развитието на женски псевдохермафродитизъм (кариотип XX).
Видео: Лечение на безплодие в Новосибирск - Начини (Част 2)
Липса на ензими, участва в синтеза на тестостерон, или AR дефицит води до мъжки псевдохермафродитизъм (кариотип XY).
олигоспермия (Концентрация на сперматозоиди на по-малко от 5 милиона / мл) или азооспермия, свързани с хромозомни аномалии в 3-13% от случаите. Тези аномалии могат да включват половите хромозоми, като например 47, 47, XXY- XYY- пол хромозомна мозаицизъм структурни аномалии Y-хромозома. За аномалии на автозоми са реципрочни транслокации, Robertsonian транслокации, инверсии и други структурни аномалии. Най-често хромозомни аномалии, свързани с не-обструктивна азооспермия, намерено синдром на Klinefelter (47, XXY).

Най-често срещаната генетична причина за вродени обструктивна азооспермия Генни мутации са трансмембранен регулатор в муковисцидоза.
синдром на Клайнфелтер - най-често срещана причина hypergonadotrophic хипогонадизъм при мъжете (високо FSH, ниско - тестостерон). Въпреки че повечето пациенти с кариотип 47, XXY, за някои характеристика мозаицизъм. В този синдром, намиране на прогресивно намаляване на популацията на зародишни клетки и стероид-фиброза и полови жлези.
клиничен проява характерни за пубертета и ненормално фенотип представени - твърде висок, малкия размер на гениталиите, необичайно окосмяване на тялото. Обикновено такива пациенти, подложени на андроген. Въпреки факта, че повечето пациенти показват, азооспермия, понякога в тестисите разкрие незрели или зрели сперматозоиди. По-голямата част на зрелия спермата, получена от пациенти за ин витро оплождане, се характеризира с нормална хаплоидна кариотип, така че тяхното потомство, също има нормален кариотип.
Микроделеции на азооспермия фактор за мъжкото безплодие. Testikulodeterminiruyuschy КРЗ ген локализира проксимално psevdoautosomnomu участък на късото рамо на Y-хромозома. На дългото рамо на региона на Y-хромозома намира три азооспермия фактор (AZF). проксималната дълго рамо, на същото място и ген Daz, са региони AZFa, AZFb и AZFc. YQ микроделеции в тези региони могат да станат причина за 10-20% от случаите на азооспермия и 3-10% от случаите на тежка олигоспермия.
Заличавания и AZFa AZFb, очевидно проявяват клинично по-тежки, отколкото AZFc заличавания, които се срещат по-често. някои мъже с YQ заличавания запазят плодовитостта. Извършва методи за лечение на изкуствено оплождане и ITSIS. Въпреки това, мъжките потомци, родени на бащите с YQ микроделеции, също е вероятно да бъде безплодно, тъй като наследи от същия микроделеция.
 Причините за фетални аномалии. Рискът от фетални малформации
Причините за фетални аномалии. Рискът от фетални малформации Кардиоваскуларни плода. Мутации в гените на транскрипционни фактори
Кардиоваскуларни плода. Мутации в гените на транскрипционни фактори Аномалии на SOx гени и синдром TVH Holt-Орам. Фактори фибробласт растежен
Аномалии на SOx гени и синдром TVH Holt-Орам. Фактори фибробласт растежен Вродена скъсяване на бедрената кост. Отцепването на ръцете и краката на плода
Вродена скъсяване на бедрената кост. Отцепването на ръцете и краката на плода Синдром на катран в плода. AASE синдром, Holt-Орам плода
Синдром на катран в плода. AASE синдром, Holt-Орам плода Намаляване на дефекти на крайниците на плода. Фокомелия плода
Намаляване на дефекти на крайниците на плода. Фокомелия плода Асоциация Vater в плода. Полидактилия синдром и фетален goldenhara
Асоциация Vater в плода. Полидактилия синдром и фетален goldenhara Синдром на Холт Орам. синдром Gidroletalny
Синдром на Холт Орам. синдром Gidroletalny Синдром frinsa. Диагностика и управление на синдрома на плода frinsa
Синдром frinsa. Диагностика и управление на синдрома на плода frinsa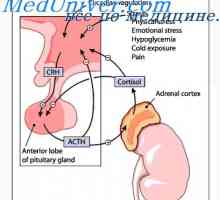 Синдром Pfeiffer. Диагноза и прогноза на синдром на Пфайфър
Синдром Pfeiffer. Диагноза и прогноза на синдром на Пфайфър Системата на мъжкия генитален тракт на ембриона. Женската полова система плода въздуховоди
Системата на мъжкия генитален тракт на ембриона. Женската полова система плода въздуховоди Понижаването на фетални яйчниците. Основите на половите органи на ембриона
Понижаването на фетални яйчниците. Основите на половите органи на ембриона Аномалии на женските полови органи. Синдроми Kaufman-мак-Cusick и Mayer-Rokitansky-Кустер-Hauser
Аномалии на женските полови органи. Синдроми Kaufman-мак-Cusick и Mayer-Rokitansky-Кустер-Hauser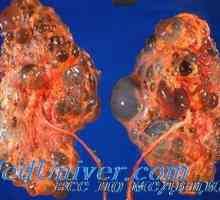 Автозомно-доминантно поликистоза на бъбреците при деца. Диагностика и лечение
Автозомно-доминантно поликистоза на бъбреците при деца. Диагностика и лечение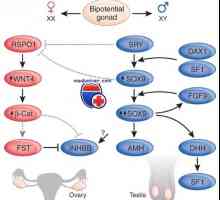 Мутация и ген дублиране dax1, sox9. пол генотип XY несъответствие и kampomelicheskaya дисплазия
Мутация и ген дублиране dax1, sox9. пол генотип XY несъответствие и kampomelicheskaya дисплазия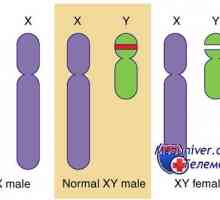 Генетични заболявания на половите жлези. Гените КРЗ, WT1 и синдроми Фрейзър и Денис-dresha
Генетични заболявания на половите жлези. Гените КРЗ, WT1 и синдроми Фрейзър и Денис-dresha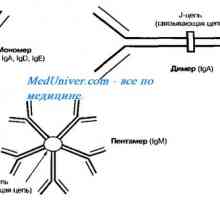 Автозомно рецесивен синдром хиперпродукция на имуноглобулин М (IgM). генна мутация помощ
Автозомно рецесивен синдром хиперпродукция на имуноглобулин М (IgM). генна мутация помощ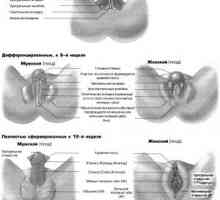 Развитието на половите органи при плода от седмица
Развитието на половите органи при плода от седмица Мюлеров аномалия
Мюлеров аномалия Хипоспадия, лечение и причини
Хипоспадия, лечение и причини Моногенни синдроми mvpr
Моногенни синдроми mvpr