Патогенезата на ембриогенезата заболяване, морфогенеза на Hirschsprung
Най-интензивно изследван аспект на дебелото черво - механизмът на възникване болест на Hirschsprung, който се характеризира с липса на вътрешни клетки парасимпатиковата ганглии, които са разположени в субмукозно плексус (Meissner) и в intermuscular плексус (Auerbach) задните черва. Развитие на заболяване е свързано с липсата на невралната тръба клетки мигрират към дисталната чревна тръба, и клинично заболяване е най-честата форма на вродена запушване на червата.
Проучване на модел мутант мишки, възможно да се обясни причината за заболяването. В изследването на мишки с присъствие на смъртоносни мутации крайните дистална чревна тръба се характеризира с вродена липса отделя ганглии. Липсата на ганглии в стената на дебелото черво е резултат от неспособността на мигриращи невралната тръба клетки да проникнат в необичайно сгъсти базално плоча и развива нормално колонизират червата тръбата. Ламинин взаимодейства с базални плоча рецептори невралната тръба клетки и спира началото на миграцията на.
по различен мутантни щамове мишки Това показва, че вроден мегаколон в трансгенни мишки, свръхекспресиращи hoxa-4, вероятно възниква поради анормално взаимодействие между прекурсорите на ентерични неврони и гладкомускулни клетки.
Генетична и молекулно машини, основното заболяване, са обект на интензивно изучаване, както е отразено в огромен брой публикации, броят на които непрекъснато се увеличава. Идентифицирани множество мутации, при наличието на които се дължи на развитието на болестта на Hirschsprung. Намерено мутации на кодиращи региони, включително т.нар точка миссенс и безсмислени мутации и делеции, което предполага нарушение на функциите на протеини, получени в резултат на гени. генни мутации са изследвани най-интензивно, кодираща тирозин киназа рецептор за (задържане ген) рецептор и ендотелин-Б.
Ret ген е protooncogen, кодира трансмембранен тирозин киназа рецептор на. Лиганда за рецептора е белязано невротрофичен фактор, произведен от глиални клетки (GDNF), изразени в развиващия червата тръбата. Сега е установено, че GDNF също се свързва към друг рецептор - GDNFRa - и двете молекули заедно образуват сигнален комплекс с Ret протеин. Хомозиготните мишки с Ret анормални протеинови продукти, характеризиращи се с недостиг киназа, необходими за образуване на ентерично ганглии. При пациенти с болест на Hirschsprung разкри наличието на множество мутации в гена на задържане. Подробен преглед на структурата и функцията на рецептора Ret-публикувани Manie и др ..

насочена увреждане ген, кодираща рецептор за ендотелин-B, придружен от създаването на мишки с липса на мегаколон и ганглии, което предполага важна роля на този рецептор, включително процесите на миграция и диференциация на невралната тръба клетки, които са оформени и ентерично ганглии. В допълнение, дефекти Ret-рецептори и рецептори за дефицит на ендотелин-B GDNF лиганди и ендотелин-3, както и ензимен дефицит на ендотелин конвертиращ, доведе до фенотип на образуване на мишки, подобни на болест на Hirschsprung. Независимо от комплексите описано рецептор / лиганд при пациенти с мутация болест на Hirschsprung се открива фактор Sox-10 - транскрипционен фактор, експресиран в клетки от естествен гребен по време на ранното развитие.
За разлика от по-рано използвани лабораторни модели на последните експериментални проучвания, проведени върху мишки, които нямат Ret-рецептори поради мутации, които имат преобладаващо отрицателен ефект. В резултат на това тези мишки са фенотипните копие от заболяване на съответния Hirschsprung и при хората. В момента лицето идентифицирани 11 гена болест на Hirschsprung, което е важно в развитието на заболяването не е същото, както и няколко други гени идентифицирани в мишки, в които мутациите са придружени от развитието на болестта, заболяването на Hirschsprung идентични.
Основният фактор, водещ до развитие на болест на Hirschsprung, - задържане генна мутация. Тази мутация е намерена при честота до 50% в случай на наследствена форма на болестта и само 20% - в спорадични изпълнение. Напоследък беше показано, че мутация в гена на регулиране фрагмент задържане също влияе на податливостта към въпросната болест.
Видео: момичетата да изглеждат до края!
анализ на задържане генен локус последователност възможно да се установи наличието на полиморфизъм, свързан с развитието на болестта и локализирана в консервативната област отговорен за свързване на транскрипционен фактор и вероятно участва в регулацията на транскрипцията. Подробно описание на сегашното състояние на научните изследвания върху генетичните основи на болест на Hirschsprung може да бъде намерен в базата данни онлайн Закони на Мендел в база данни Man (Закони на Мендел в Ман).
Определяне на стойността на взаимосвързани процеси пролиферация, миграцията и функция на невралната тръба клетки е една от стъпките в разбирането на механизмите на развитие на болест на Hirschsprung. Изследването на изолирани невронни гребен стволови клетки, използвайки модел лабораторни мишки е началната точка в обяснява някои от процесите. Iwashita и сътр. изолирани невронни гребен стволови клетки от ембрионален червата на плъхове и изследвани генната експресия с помощта на микрочип технология. Учените са открили, че предварително идентифицирани гени, свързани с развитието на болест на Hirschsprung бяха в стволовите клетки на невралната тръба активиран.
Резултатите потвърждават методи полимеразна верижна реакция обратна транскрипция, поточна цитометрия и функционален анализ. В ин витро експерименти показа, че GDNF насърчава клетъчна миграция, но няма ефект върху клетъчната пролиферация и клетъчната жизнеспособност. Данните показват също, взаимодействието на сигнални пътища GDNF и ендотелин в регулацията на миграцията на невронни стволови клетки гребен. Способността на стволови клетки в невронната гребен терапия е проучен в експерименти, в които е доказано ефективна интеграция на трансплантираните клетки в червата на мишки с ендотелин рецепторни дефекти и Ret-рецептори.
Текущо инсталирани основните регулатори на храносмилателния тракт, както и някои гени, необходими за епителни-мезенхимни взаимодействия - основните механизми на ембрионалното развитие. Анализ на генната експресия модели кутия предполага тяхното участие във формирането на храносмилателния тракт. Семейни таралеж протеини влияят няколко аспекта на формирането на храносмилателния тракт по време на ранното развитие на плода. Насочени заличаване на отделни гени, участващи в образуването на язви, дължащи се на създаване на мутирали животни разкрива важна регулаторна роля на ВМР в производствените процеси на клетъчна пролиферация, морфология характерен вид въси и крипти позициониране. Допълнително проучване на гени, включени в регулирането на механизми гастроинтестинални развитие следва да се улесни по-доброто разбиране на патогенезата на процеси и нови подходи за лечение на стомашно-чревни заболявания.

 Uzi дебелина фетален черво. Аноректалното малформации при плода
Uzi дебелина фетален черво. Аноректалното малформации при плода Ganglinarnaya плоча ембрион. Основни мозъчни везикули
Ganglinarnaya плоча ембрион. Основни мозъчни везикули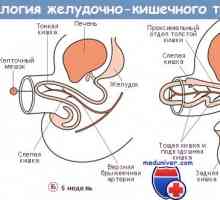 Получаване на червата. Развитие на плода черво
Получаване на червата. Развитие на плода черво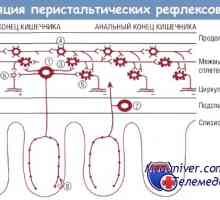 Медиатори ентерични неврони. Парасимпатиковата и симпатичен инервацията на червата
Медиатори ентерични неврони. Парасимпатиковата и симпатичен инервацията на червата- Ентералната нервна система. Intermuscular и субмикозен сплит
 Възвратно-постъпателно движение (перисталтиката) на червата. Регулиране на движението на червата
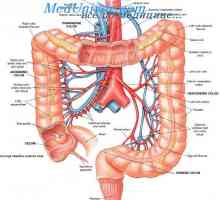
Възвратно-постъпателно движение (перисталтиката) на червата. Регулиране на движението на червата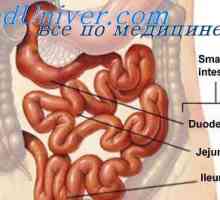 Повишена перисталтика на тънките черва. Физиология на илеоцекалната клапа
Повишена перисталтика на тънките черва. Физиология на илеоцекалната клапа Миграцията нарушения на ентерично нервна система при болест на Hirschsprung
Миграцията нарушения на ентерично нервна система при болест на Hirschsprung- Физиология на ентеричното нервна система
 Синдром на късото черво (CCM) причинява, епидемиология
Синдром на късото черво (CCM) причинява, епидемиология Хормонът контролира рак на червата
Хормонът контролира рак на червата- Болест на Hirschsprung информация за пациента
- Megakolongigantizm дебелото черво различен произход (болест на Hirschsprung, болест на Chagas,…
- Механично чревна обструкция е обструктивно и задушаване. Когато обструктивно илеус има компресия…
- Здраве Енциклопедия, болест, лекарства, лекар, аптека, инфекция, резюмета, пол, гинекология,…
- Здраве Енциклопедия, болест, лекарства, лекар, аптека, инфекция, резюмета, пол, гинекология,…
- "Играя догонване": механизъм движение клетка обяснява образуването на метастази
 Мозъчния рак на щитовидната жлеза
Мозъчния рак на щитовидната жлеза Болест на Hirschsprung
Болест на Hirschsprung Хронична или частично запушване на червата
Хронична или частично запушване на червата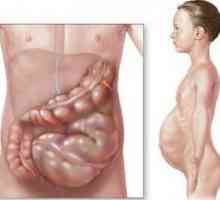 Мегаколон, токсичен: Лечение, Симптоми
Мегаколон, токсичен: Лечение, Симптоми