Вродена и наследствено тубулопатия при деца
Видео: Причини за Аутизъм: генетика, ген за аутизъм, ваксина DTP, глутен
Съдържание

Тубулопатия - заболяване с първични лезии водещ тръбна в която счупен мембранен транспорт на различни вещества в бъбречните каналчета.
Видео: Инстинкти, деца, стрес, образование - Жак Фреско - Проектът Венера
При новородени открити клинично изолиран тубулопатия, които са най-вече придружени от полиурия и нарушаване на електролити в плазмените нива. Те включват синдром на Бартер, Liddle, Лоу, безвкусен диабет, първичен тип gipoaldosteronizm I, хиперкалцемия от "ранно детство" или тръбен тип ацидоза IV. Освен синдром на Бартер и Lowe, морфологични промени в бъбреците при тези тубулопатия отсъства или се характеризира с развитието на нефрокалциноза.
Видео: диагностика на вродени и наследствени патология Семинар
синдром на Бартер - тубулопатия, хипокалемия проявява, chloropenia, метаболитна алкалоза, и хипер-reninemiey с нормално кръвно налягане. Има три форми на синдрома: неонатална, класически и синдром на Gitelman (при новородени не се среща). Новородени и класически синдроми Бартер понякога наследени автозомно рецесивни, но повечето случаи на спорадично. Идентифицирани от две генотип подвидови неонатални форми на синдрома. Подтип I, причинени от мутации в гена kotransportnom на натриев и калиев хлорид (SLC12A1 локус на дългото рамо на хромозома 15-15q15-21), подтип II - ROMK мутация в гена (KCNJ1 локус на дългото рамо на хромозома 11 (11q24-25) За класически синдром на Бартер. характеризираща се с мутации в гена на натриев канал (локус CLCNKB на късото рамо на хромозома 1-1r36). в двете форми съществуват Polyhydramnios майки и бебета често са преждевременно родени, с раждането може да бъде масивна полиурия. в изследването на околоплодна течност във зародишен период се открива SW lichenie съдържание хлорид, както е определено чрез ехография нефрокалциноза, хидронефроза и понякога в резултат на хронични хидроуретер полиурия.
В неонатална форма се диагностицира скоро след раждането, класическа - при новородени или кърмачета на възраст до две години.
Видео: Вродена болест на децата - е предизвикателство за родителите?
Микроскопски наблюдава, заедно с хиперпластични нефрокалциноза юкстагломеруларния апарат (патогномонична знак), по-рядко - хиперплазия, медуларни интерстициални клетки, понякога - hyalinosis гломерулна вакуолизация апикални епителни тубули, тръбна атрофия и интерстициална фиброза. Смъртността и смъртност се наблюдава в нетретирана синдром на Бартер. При лечението на състоянието на детето значително се е подобрило, но в дългосрочен план прогноза остава несигурна, тъй като постепенно прогресира бавно CRF.
синдром Lowe (oculocerebrorenal синдром) - рядко генетично заболяване е Х-свързана рецесивен режим на наследяване. Синдромът е причинена от генна мутация OCRL1 разположен на X хромозомата (Xq25-26). Жените - носители на анормален гена имат катаракта, но те имат нормална бъбречна и неврологичната функция. Заболяването се характеризира с комбинация от вродени катаракта или глаукома, тежко умствено изоставане, хипотония с тубулна дисфункция.
Интензивност и време на тръбна дисфункция са променливи. Бъбречната функция и структура при раждането може да е нормална. Каналчета дисфункция често започва на възраст между 3-12 месеца между тях. Микроскопични структура на бъбреците при фетуси и новородени обикновено е нормално. Въпреки това, още в първите месеци от живота се наблюдава разширяване на тубулите, те атрофират и протеинови цилиндри в лумена. Гломерули от нормални новородени и след това те създават склероза (фокална или глобална гломерулосклероза), GBM сгъсти. Електронна микроскопия посочено крака сливане podocytes и тръбна митохондриално увреждане, но не е ясно дали са първични във връзка с тръбната дисфункция.
 Вродена идиопатична хиперкалцемия и hypophosphatasia. крехки кости
Вродена идиопатична хиперкалцемия и hypophosphatasia. крехки кости Галактоземия
Галактоземия Метаболитна алкалоза и първа помощ
Метаболитна алкалоза и първа помощ- Спешна помощ в хипокалиемия
- Първа помощ при хипо- и chloruremia
- Аутизъм научат да идентифицират деца на шест месеца
 Диагностика и диференциране на синдром на Кон. Лечение на първичен алдостеронизъм
Диагностика и диференциране на синдром на Кон. Лечение на първичен алдостеронизъм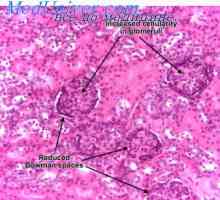 Лезиите в бъбречните тубули. Глюкозурия, acidaminuria, фосфатурия
Лезиите в бъбречните тубули. Глюкозурия, acidaminuria, фосфатурия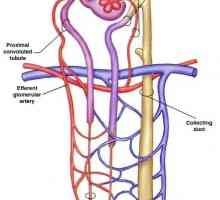 Distalnokanaltsevy ацидоза при деца. клиника
Distalnokanaltsevy ацидоза при деца. клиника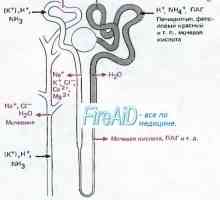 Бъбречно тръбна ацидоза. Proksimalnokanaltsevy
Бъбречно тръбна ацидоза. Proksimalnokanaltsevy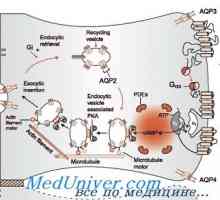 Рахит с тръбна ацидоза. Нефрогенна безвкусен диабет при деца
Рахит с тръбна ацидоза. Нефрогенна безвкусен диабет при деца- Синдром на Gitelman в децата. Диагностика и лечение
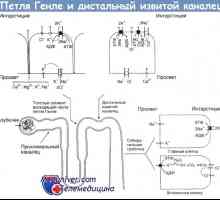 Синдром на бартер при деца. Диагностика и лечение
Синдром на бартер при деца. Диагностика и лечение- В Америка, епидемията от аутизъм
- Безвкусен диабет
- Етиологията и патогенезата на хиперкалциемичния криза
- Бъбречно заболяване терапия,
 Pseudohyperaldosteronism
Pseudohyperaldosteronism Бъбречно тръбна ацидоза: симптоми, причини, лечение, симптоми
Бъбречно тръбна ацидоза: симптоми, причини, лечение, симптоми Други патология при новородени
Други патология при новородени Вродена и наследствено гломерулопатия
Вродена и наследствено гломерулопатия