Нарушения на метаболизма на мастни киселини и глицерол

Съдържание
По време на пост, по-голямата част от енергията на тялото трябва да се реализира за сметка на метаболизма на мазнините.
Използването на мазнини като източник на енергия катаболизма на мастната тъкан трябва да свободни мастни киселини и глицерол. Свободните мастни киселини се метаболизира в черния дроб и периферните тъкани чрез окисление на ацетил-СоА в черния дроб глицерол, използвани в синтеза на триглицериди или глюконеогенеза. Основни нарушения - карнитин, но вторичен дефицит на карнитин е вторично биохимично проява на много органични acidemia и дефекти на окислението на мастни киселини.
Нарушения -oksidaznogo цикъл
Тези процеси се характеризират с множество генетични дефекти, които обикновено се появяват по време на хипогликемия на гладно и atsidozom- някои причина кардиомиопатия и мускулна слабост.
Видео: липидния метаболизъм - Част 2
Ацетил-СоА се генерира от мастните киселини от повтарящи се цикли -oksidaznyh. Комплект от четири ензими (atsildegidrogenaza, хидратаза и hydroxyacyl дехидрогеназа лиаза), специфични за различни дължини на веригите (много дълги вериги, с дълга верига, средна верига и къса верига), необходими за пълно katabolizirovaniya дълговерижни мастни киселини.
Недостигът на среда-ацил дехидрогеназа (MCADD). Този дефицит е най-честият дефект -oksidaznogo цикъл и бе включен в разширената неонатален скрининг в много държави.
Клиничните прояви обикновено се появяват след 2-3-месечна възраст и обикновено следват един епизод на гладно (поне 12 часа). При пациенти, които имат повръщане и сънливост, която може бързо да прогресира до припадъци, кома и понякога смърт (което може да се прояви и като внезапната детска смърт). По време на атаки, пациентите имат хипогликемия, хиперамонемия и неочаквано ниски нива на кетони в урината и серума. Често има метаболитна ацидоза, но по-късно може да се прояви.
Диагнозата се установи чрез определяне средна верига конюгати на мастна киселина с карнитин в плазмата или в урината глицин или чрез откриване ензим дефицит в култура, но fibroblastov- анализ на ДНК може да потвърди, по-голямата част от случаите.
Лечение на остри пристъпи на интравенозно приложение на 10% разтвор на декстроза, 1,5 пъти нивото който поддържа zhidkosti- някои лекари също препоръчват добавянето карнитин по време на остри епизоди. Профилактика включва прилагането на диета с ниско съдържание на мазнини и високо - въглехидрати и избягване на продължителното гладуване. Царевично нишесте терапия често се използва за осигуряване на свобода на безопасност по време на гладуване през нощта.
Липса на дълга верига hydroxyacyl-СоА дехидрогеназа (LCHADD). Този недостатък е вторият най-често дефект в окислението на мастни киселини. Той споделя много функции MCADD, но пациентите могат да имат kardiomiopatii- рабдомиолиза, значително повишаване на креатин киназа и миоглобинурия време мускулна napryazhenii- периферна невропатия и чернодробна дисфункция. Майка, бременна с плодове LCHADD, често имат по-HELLP синдром по време на бременност.
Диагнозата се основава на откриването на излишък от дълговерижни хидрокси киселини в анализа на органичната киселина и в присъствието на техните конюгати с atsilkarnitinovom профил карнитин или глицинови конюгати atsilglitsinovom профил. LCHADD може да се потвърди чрез изследване на ензими в кожни фибробласти.
Лечението включва обостряния по време на хидратация, високи дози глюкоза, залежаване, алкализиране на урината и карнитин. Продължително лечение включва диета с високо въглехидратно добавка е средноверижни триглицериди и избягване на глад и упражнения верига.
Липса на много дълга верига ацил-СоА дехидрогеназа (VLCADD). Този дефицит е подобен на LCHADD, но обикновено се свързва със значително кардиомиопатия.
Глутарова acidemia тип II. Дефект в електронен трансфер от мастни ацил СоА дехидрогенази за електронна транспортна верига вредни реакции, включващи мастни киселини с верига dliny- всяко окисление на няколко аминокиселини също са засегнати.
Така Клиничните прояви включват хипогликемия на гладно, тежка метаболитна ацидоза и хиперамонемия.
Диагнозата се основава на увеличението ethylmalonic, глутарова киселина, 2- или 3-droksiglutarovoy пръти и други дикарбоксилни киселини в анализа на органична киселина, и глутарилацилаза, изовалерил и други ацилкарнитини в проучвания с тандемна мас спектрометрия. Липсата на ензими в кожните фибробласти може да бъде потвърдително знак.
Лечението е подобен на този за MCADD, с изключение на рибофлавин може да бъде ефективен при някои пациенти.
Нарушенията на глицерол метаболизъм
Глицерол се превръща в глицерол-3-фос-конте глицерол kinazoy- чернодробните ензими дефицит води до епизодични повръщане, летаргия и хипотония.
Глицерол киназа дефицит X stseplen- много пациенти с този недостатък са също хромозомни делеции, които се простират отвъд ген глицерол киназа в съседна област гени, съдържащи гени, вродена надбъбречна хипоплазия и мускулна дистрофия на Дюшен. По този начин, пациенти с дефицит на глицерол киназа могат да имат един или повече от тези заболявания.
Симптомите започват да се появяват на всяка възраст и обикновено се придружават от ацидоза, хипогликемия и високо съдържание на глицерол в кръвта и урината.
Диагнозата е откриване на повишени нива на глицерол в серума и урината и потвърдени чрез ДНК анализ.
Лечението се състои от диета с ниско съдържание на мазнини, но при пациенти с хипоплазия на надбъбречната Заместителното лечение с глюкокортикоиди е от решаващо значение.
 Първа помощ в диабетна кетоацидоза: патогенезата на заболяването
Първа помощ в диабетна кетоацидоза: патогенезата на заболяването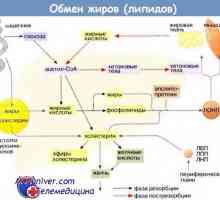 Метаболизма на мазнините в тялото. Транспорт на липидите
Метаболизма на мазнините в тялото. Транспорт на липидите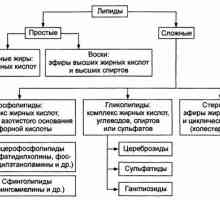 Свободни мастни киселини. Транспорт свободни мастни киселини
Свободни мастни киселини. Транспорт свободни мастни киселини- Метаболизма на мазнините и отлагане. мазнини черен дроб
- Липопротеини. Форми и физиологията на липопротеини
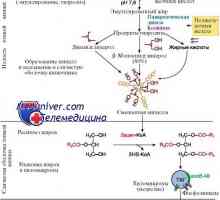 Образованието в черния дроб ацетоацетат. Кетоза време на гладно и пристрастяване към мазни храни
Образованието в черния дроб ацетоацетат. Кетоза време на гладно и пристрастяване към мазни храни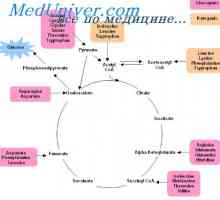 Синтез на триглицериди от въглехидрати. Етапи на синтеза на мазнини от въглехидрати
Синтез на триглицериди от въглехидрати. Етапи на синтеза на мазнини от въглехидрати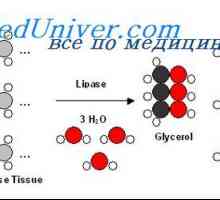 Триглицеридите синтеза на протеини. Регламент на енергия освобождаване на триглицеридите
Триглицеридите синтеза на протеини. Регламент на енергия освобождаване на триглицеридите- Хормонални да регулира метаболизма на мазнините. прекалена пълнота
 Чернодробна система на макрофагите. Метаболитен чернодробна функция
Чернодробна система на макрофагите. Метаболитен чернодробна функция Ролята на растежен хормон в метаболизма на мазнините. Въглехидратен метаболизъм и растежен хормон
Ролята на растежен хормон в метаболизма на мазнините. Въглехидратен метаболизъм и растежен хормон Ефект на кортизол на протеиновия метаболизъм. Кортизолът и метаболизма на мазнините
Ефект на кортизол на протеиновия метаболизъм. Кортизолът и метаболизма на мазнините Инсулин и глюкоза на мозъка. Ефект на инсулин върху метаболизма на мазнините
Инсулин и глюкоза на мозъка. Ефект на инсулин върху метаболизма на мазнините Причини за възникване на кетоза и ацидоза. Ефект на инсулин на протеин оборот
Причини за възникване на кетоза и ацидоза. Ефект на инсулин на протеин оборот Влиянието на хипофизата в метаболизма на мазнините. Атеросклерозата в разстройства на…
Влиянието на хипофизата в метаболизма на мазнините. Атеросклерозата в разстройства на… Характеристики на мазнини (липиди) и мастни киселини
Характеристики на мазнини (липиди) и мастни киселини Методи за оценка на метаболизма на въглехидрати и мазнини в тялото
Методи за оценка на метаболизма на въглехидрати и мазнини в тялото- Механизмите на храносмилането и усвояването на мазнини (липиди)
 Нужди на мазнини (липиди) в много недоносени новородени
Нужди на мазнини (липиди) в много недоносени новородени- Омега-3 мастни киселини може да помогне при лечението на психични заболявания
- Използването на полиненаситени мастни киселини, по време на бременност влияе на телесното тегло за…