Х-свързана тежка комбинирана имунна недостатъчност момчета. Имунна недостатъчност с аденозиндеаминаза
Х-свързана тежка комбинирана имунна недостатъчност (TKIDH1) - най-често под формата на САЩ тежък комбиниран имунодефицит, което представлява около 45% от всички случаи. Тази форма се различава от други синдром (поради дефект, различна от Jak3) постоянно нисък процент на Т- и NK-клетки и увеличава процента на В-клетки (Т ~, B +, NK). В основата на Х-свързана тежка комбинирана имунна недостатъчност лежат мутации локализирани на X хромозомата (ql3 част) и кодираща общ за рецепторите на цитокини (IL-2, IL-4, IL-7, IL-9, IL-15 и IL -21) у верига (YC).
Тази схема увеличава афинитета рецептори към съответните цитокини, осигуряване на сигнална трансдукция в клетките. Тези цитокини играят критична роля в узряването на клетките на имунната система. Следователно, когато дефекти рецепторни мустаци съединение се появява тежка имунна недостатъчност. В първите 136 пациенти изследвани бяха идентифицирани 95 различни мутации в екзон 8 на гена, кодиращ IL-2 рецептора. Повечето случаи са включени само един или няколко нуклеотида.
В 2/3 от тези мутации са установили, синтез YC дефектна верига, в друга - води до пълното му отсъствие. Носители дефект открити неслучайно Х-хромозома инактивиране или вредни мутации в Т, В или NK-клетките. Ефективността на трансплантация на костен мозък зависи от развитието в тялото на пациента не само T, но също така и В- и NK-клетките на донора.
автозомно-рецесивно наследяване тежка комбинирана имунна недостатъчност у нас е по-малко общи неща, отколкото в Европа. Когато този синдром идентифицирани мутации 7 на автозомно гени, което води до дефицит на ADA, Jak3, а-веригата на IL-7 (K7I-7Ra) рецептор 1 или 2 рекомбинази (RAG1 или RAG2), Artemis и CD45 протеини.

Имунна недостатъчност с аденозиндеаминаза
Приблизително 15% пациенти тежък комбиниран имунодефицит, липсва ензим аденозин деаминаза (ADA). В основата на този недостатък са различни точкови мутации или делеции на части от гена, разположен на хромозома 20 (ql3-трет част). Натрупване на големи количества аденозин, 2'-деоксиаденозин и 2'-О-метил аденозин, пряко или косвено води до апоптоза на Т-лимфоцити.
При пациенти с този дефицит ензим лимфопения, обикновено се изразява в много по-голяма степен, отколкото в други форми на SCID. Абсолютният брой на лимфоцитите в средната достига 500 до 1 л, като по този начин значително се намалява, тъй като броят на Т- и В- и NK-клетки. NK-клетъчната функция е напълно запазена. Трансплантация на костен мозък (не преди химиотерапия) възстановява функцията на Т-лимфоцити и нормализира функцията на В-лимфоцити.
По-леките форми на неуспех АДА (ADSS) обикновено се диагностицира по-късна възраст, а понякога дори и възрастни. Характеристиките на тази форма на SCID аномалии включват също ребра наподобяващи рахитичен "гранули" и други костни структури (издатини преимуществено илиачна кост и прешлени).
Както и при други форми на тежка комбинирана имунодефицит, в тези случаи се използва HLA-идентични трансплантации или haploidentical костен мозък изчерпани Т-лимфоцити, без преди или след химиотерапия. В такива случаи ензим-заместителна терапия не се извършва, тъй като това заплашва да отхвърляне на присадка. Генна терапия не дава резултати са обнадеждаващи. Случай на спонтанно възстановяване на функциите на повреден ген.
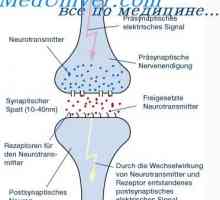 Тежък комбиниран имунодефицит. Морфология алимфоплазия на тимусната жлеза
Тежък комбиниран имунодефицит. Морфология алимфоплазия на тимусната жлеза Имунодефицит с ахондроплазия. Морфология имунодефицитен с ахондроплазия
Имунодефицит с ахондроплазия. Морфология имунодефицитен с ахондроплазия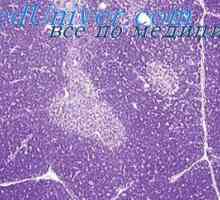 Комбиниран имунодефицит с fermentopathy. синдром nezelofa или alymphocytosis
Комбиниран имунодефицит с fermentopathy. синдром nezelofa или alymphocytosis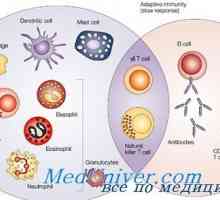 Вродения имунитет. Модерният идеята за вродения имунитет
Вродения имунитет. Модерният идеята за вродения имунитет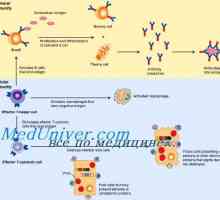 Активирането на вродения имунитет. Етап активирането на вродения имунитет
Активирането на вродения имунитет. Етап активирането на вродения имунитет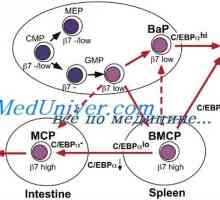 Диференциацията на Т и В клетки. Цитокините индуцират диференциация на клетки от Th1 тип и от типа…
Диференциацията на Т и В клетки. Цитокините индуцират диференциация на клетки от Th1 тип и от типа…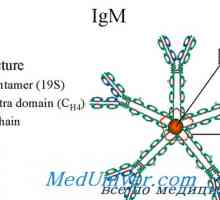 Х-свързан синдром хиперпродукция на имуноглобулин М (IgM) момчета. Мутация на CD40 CD154
Х-свързан синдром хиперпродукция на имуноглобулин М (IgM) момчета. Мутация на CD40 CD154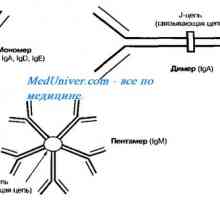 Автозомно рецесивен синдром хиперпродукция на имуноглобулин М (IgM). генна мутация помощ
Автозомно рецесивен синдром хиперпродукция на имуноглобулин М (IgM). генна мутация помощ Тежка комбинирана имунна недостатъчност при деца. Симптомите и лечението
Тежка комбинирана имунна недостатъчност при деца. Симптомите и лечението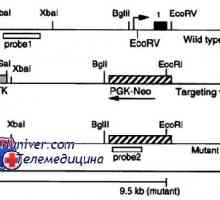 Мутации гонадотропин рецептори. Аномалии на LH и FSH рецептори
Мутации гонадотропин рецептори. Аномалии на LH и FSH рецептори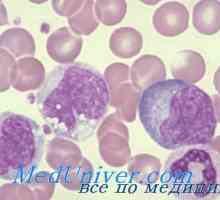 Нарушенията на Т-лимфоцити. CD8 лимфопения
Нарушенията на Т-лимфоцити. CD8 лимфопения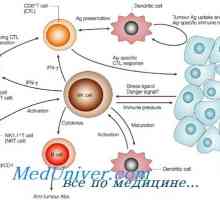 Образуване на NK-фетален имунни клетки. Т-лимфоцитна функция имунитет
Образуване на NK-фетален имунни клетки. Т-лимфоцитна функция имунитет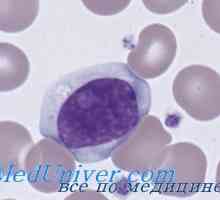 Имунодефицити в недостатъчност JAK3, ил-7ra, rag1 или rag2, CD45
Имунодефицити в недостатъчност JAK3, ил-7ra, rag1 или rag2, CD45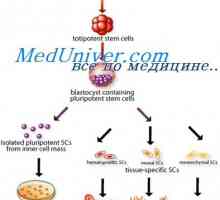 Трансплантация на стволови клетки в имунната недостатъчност и анемия на Fanconi
Трансплантация на стволови клетки в имунната недостатъчност и анемия на Fanconi- Ваксина срещу ХИВ може да се появи след две години
- Мъжете с по-високи нива на имунитет привлекателни за жените
- В Европа научихме за лечение на тежка комбинирана имунна недостатъчност
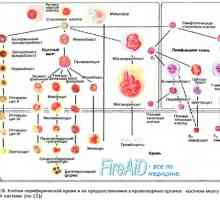 Т-лимфоцитна функция. Активираните Т-лимфоцити. Цитокини.
Т-лимфоцитна функция. Активираните Т-лимфоцити. Цитокини.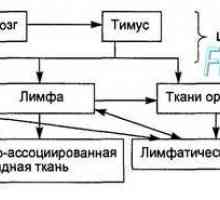 Имунната система. Човешката имунна система. Non-специфичен имунитет. Специфичен имунен отговор.…
Имунната система. Човешката имунна система. Non-специфичен имунитет. Специфичен имунен отговор.…- Имунологична толерантност. Механизми контролират имунната система. Хормонални контрол на имунната…
- Терапия