Метаболитни нарушения на цикъла на пикочна киселина

Клиничните прояви на тази разнородна група LVP, че вероятно е свързано със степента на остатъчна ензимна дейност.
Разграничаване форми с неонатална. инфантилен или повече късно начало на заболяването. Клиничните прояви могат да бъдат вече в неонаталния период и включват хипотензия, сънливост, конвулсии, кома. Продължаващо открива хиперамонемия в комбинация с дихателна алкалоза.
Недостигът ornitintranskarbamilazy - един от най-често срещаните форми на патология, има Х-свързана рецесивен наследяване. Генът карти за Hr21.1. Хомозиготни момчета в първите три дни на живота разработен повръщане, прогресивна летаргия. Промените са неспецифични: хепатомегалия с фокална некроза и мастна дегенерация. Лица, които са хетерозиготни за по-благоприятно, в черния дроб разкрити свързващи некроза, възпалителна реакция, фиброза и стеатоза. Електронна микроскопия - набъбване и освобождава пероксизомен матрица. В структурата на митохондриите не е счупена. В типа -appearance CNS II астроцитите на Алцхаймер хетеротопия продълговатия мозък, спонгиозност и демиелинизация.
Karbamilfosfatsintetazy дефицит е автозомно-рецесивно модел на унаследяване. Генът карти за 2q35. болест на мълния, възрастта на настъпване на болестта - неонаталния период. В черния дроб - слабо маркирана стеатоза, некроза на хепатоцитите, умерена портал фиброза. Възможни са промени на митохондриите: подуване, плътни депозити в матрицата, нарушена Christie. В централната нервна система - спонгиоза, появата на астроцитите тип II на АД.
Tsitrulinemiya - argininoyantarnoy киселина дефицит синтетаза. Мутации в структурния ген цитозолни argininoyantarnoy киселина синтетаза. Генът карти за 9d34. Клинични прояви и морфологични промени са същите, както в други нарушения на метаболизма на пикочната киселина. Характерно пулверизиране мастен черен дроб, синдром на Reye наподобяващи.
Синдром Hyperornithinemia--хиперамонемия-gomotsitrullinemii - основната Биохимичният дефект е неизвестен. Генът карти за 13q34. Intermitgiruyuschee заболяването протича с различна начална дата. Това може да се случи в неонаталния период когато изкуствено хранене като реакция на протеинови храни с повръщане, сънливост, конвулсии. При хронична Разбира се случва атаксия и хореоатетоза, умствена и физическа изостаналост. Електронна микроскопия в черния дроб, скелетен мускул, бели кръвни клетки, открити патология на митохондриите: удължение, полиморфизъм, присъствието на кристалоиди.
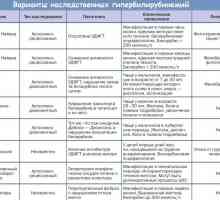 Синдром Zhilbera- lerebulle фамилна хипербилирубинемия. синдром Crigler - Najjar и lyutseya - Arias…
Синдром Zhilbera- lerebulle фамилна хипербилирубинемия. синдром Crigler - Najjar и lyutseya - Arias… Nemaline миопатия. митохондриална миопатия
Nemaline миопатия. митохондриална миопатия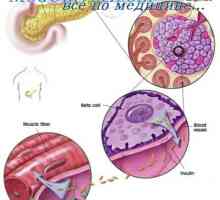 Автозомно-рецесивно заболяване. Х-свързана наследство
Автозомно-рецесивно заболяване. Х-свързана наследство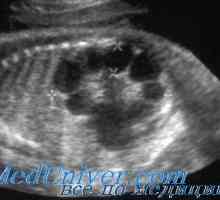 Автозомно-рецесивно поликистоза на бъбреците при деца. Диагностика и лечение
Автозомно-рецесивно поликистоза на бъбреците при деца. Диагностика и лечение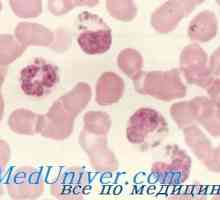 Липса на миелопероксидаза (МРО). Клиника и диагностика
Липса на миелопероксидаза (МРО). Клиника и диагностика- Основно митохондриална хепатопатия клиника, диагностика
 Клинична картина на меките тъкани на компресия
Клинична картина на меките тъкани на компресия- Хемофилия е наследствено заболяване, предадена от рецесивен Х-свързана тип, проявява нарушения…
 Нарушение на липидния метаболизъм
Нарушение на липидния метаболизъм Пероксизомен заболяване
Пероксизомен заболяване Заболявания, предизвикани от дефекти окисление на мастни киселини
Заболявания, предизвикани от дефекти окисление на мастни киселини Първа помощ при отравяне хипнотични лекарства
Първа помощ при отравяне хипнотични лекарства Болести метаболити транспорт
Болести метаболити транспорт Обмен нарушения метали
Обмен нарушения метали Въглехидратен метаболизъм при кърмачета
Въглехидратен метаболизъм при кърмачета Вродена и наследствено тубулопатия при деца
Вродена и наследствено тубулопатия при деца Нарушения на метаболизма на мастни киселини и глицерол
Нарушения на метаболизма на мастни киселини и глицерол Мукополизахаридоза деца, 1,2,3 вид лечение, диагностика, симптоми
Мукополизахаридоза деца, 1,2,3 вид лечение, диагностика, симптоми Метаболитни нарушения в метаболизма на пикочната киселина и метали
Метаболитни нарушения в метаболизма на пикочната киселина и метали Органични aciduria
Органични aciduria Пероксизомен разстройство
Пероксизомен разстройство