Кариотип 46 хх жена

Съдържание
Кариотип 46 XX жена.
Мъжки фенотип с кариотип 46, XX
В този случай, има сексуална диференциация на мъжки тип, въпреки кариотипът 46, XX. Това обикновено се дължи на обмена на различни части на X- и Y-хромозома на бащиния време на гаметогенезата. В 60% от тези пациенти КРЗ ген се прехвърля на късото рамо на Y-хромозома в Х-хромозомата или автозоми. В останалите пациенти КРЗ ген не е идентифициран, което включва транслокацията и активиране на други гени, отговорни за развитието на тестисите и благосъстоянието на Y-хромозома, X хромозомата или автозоми. В резултат, X хромозома на аномален бащата, свързване с нормален майката Х-хромозома, или autosome показва активиране на гени, отговорни за мъжкия полов тип диференциация. В присъствието на КРЗ генен индиферентни половите жлези превърнати в яйце с нормална ендокринната функция, която секретира анти-Мюлеров хормон и андрогени. Сперматогенезата по този начин не се случи, тъй като съответните гени са разположени на дългото рамо на Y-хромозома и не се прехвърлят към Х-хромозомата. Нормално мъжки фенотип е трудно да се диагностицира, докато пациентът не ходят на лекар с оплаквания от безплодие. Външно, тези пациенти са подобни на пациенти със синдрома Клайн-Felter изключение на растеж - с обръщане етаж 46, XX под средния ръст поради липса разположен върху Y-хромозома гени, които осигуряват растеж. При възрастните тестисите обикновено са намалени в razmerah- възможно крипторхизъм. Полово окосмяване по-близо до женски тип. Малък размер на пениса. По-малко от 10% от пациентите разкрие хипоспадия. Когато изследване на спермата разкрие азооспермия.
Вродена надбъбречна хиперплазия
Причина нарушения - дефект в стероидогенезата ензими. В САЩ и Европа, честотата на CYP21 генни мутации, кодиращи 21-хидроксилаза, е от 1 до 5000 1 на 15,000 раждания. По-малко общ недостатък 11-хидроксилаза (CYP11B1 ген) и Z-хидроксистероид дехидрогеназа (HSD3B2 ген). Дефекти в тези ензими води до значително намаляване или пълна липса на секреция на кортизол, но също така да mineralkortikoidnoy недостатъчност. Липсата инхибиторното действие на кортизол хипофизната води до увеличаване на производството на АСТН. Постоянно стимулиране с високи концентрации на надбъбречната АСТН причинява хиперплазия на жлезиста тъкан и прекомерно кортизол секрецията на андрогени и прекурсори.
Надбъбречните жлези на плода се образуват в третия месец на развитието на плода, когато яйчниците имат напълно функционираща, Wolffian канали атрофирали и диференциация Мюлеров пред завършване.
Недостигът 11-хидроксилаза
дефицит на 11-хидроксилаза е относително рядко и е около 16% от класическата форма на надбъбречна хиперплазия. С увеличаване на нива на деоксикортикостерон разработен натриев задържане и хипертония - отличителните черти на тази форма на заболяването.
Ензимните 11-хидроксилаза кодират двата гена - CYP11B1 и CYP11B2. 11 недостатъчност хидроксилаза възниква предимно когато CYP11B1 генни мутации. CYP11B2 генни мутации, открити рядко.
3-хидроксистероид дехидрогеназа дефицит
W-хидроксистероид дехидрогеназа се изразява в надбъбречните жлези и гонадите. Делът на дефицита на ензима представлява 1-10% от всички случаи на вродена надбъбречна хиперплазия. Вид на наследството - автозомно-рецесивно. Причина за болестта - HSD3B2 мутация ген, кодиращ 3-хидроксистероид дехидрогеназа тип II. За разлика от провала на 21-хидроксилаза и 11-хидроксилаза, с 3-хидроксистероид дехидрогеназа дефицит стероидогенезата разбити не само в надбъбречните жлези, но и в половите жлези. Обикновено има синдром на загуба на сол в ранна възраст и двойна структура на гениталиите от двата пола. Излишък на DHEA причинява лека хипертрофия на клитора на момичетата, но мъжествено излъчване те обикновено не се случи. Boys 3-хидроксистероид дехидрогеназа дефицит води до непълно маскулинизация на външните гениталии. Две трети от пациентите са описани кариотип 46, XY. При деца от двата пола признаци открити светлина хиперандрогенизъм (например преждевременна adrenarche) поради синтеза на андрогените в периферните тъкани под действието на тип I 3 -gidroksisteroidtsegidrogenazy когато недостатъчност 3 - gidroksistero iddegidrogenazy тип II. Наличието на три тип I -gidroksisteroidtsegidrogenazy усложнява хормонално диагностициране на заболяване, дължащо се на трансформацията на значителни количества от 17-hydroxypregnenolone в 17-хидроксипрогестерон.
В некласически форма на заболяването е много рядко. Диагностика на преждевременно adrenarche във връзка с повишени нива на 17-хидро-ksipregnenolona над 290 нмол / л (около 54 стандартни отклонения над нивото наблюдава в контролната група на юноши с срамната разпределение коса съответстващ Tanner етап 2). В момента жените в детеродна възраст с хиперандрогения и нормални полови органи не правят диагнозата на некласически форми на 3-хидроксистероид дехидрогеназа дефицит, тъй като увеличението на 17-hydroxypregnenolone тези жени, очевидно се дължи на синдром на поликистозните яйчници.
Хиперандрогения майката
андрогени плацентата (с изключение на дихидротестостерон) се превръщат в естрогени, обаче обикновено вирилизация плода не се случва дори когато значителен излишък от андрогени в тялото на майката.
ендогенни андрогени
Вирилизация на женски плода може да се случи в присъствието на майката тяло androgensekretiruyushey тумор (обикновено lyuteomy бременност) тъкан, която съдържа 5 редуктаза и секретира дихидротестостерон. Пето въглероден атом в молекула дихидротестостерон наситен vodorodom- резултат не се подлага на ароматизиране андроген и акумулира в тялото. Съществува хипотеза, че на маскулинизация на женския фетус може да се дължи на образуването на големи количества тестостерон, които не разполагат с време, за да се превърне в естроген. Въпреки това, не е получена тази доказателства. Други редки androgensekretiruyuschie яйчниците или надбъбречните тумори, причиняващи фетален вирилизация: adrenoblastomy, leydigomy или метастази на епителни тумори.
екзогенни андрогени
За лечението на някои заболявания (например ендометриоза) преди прилага синтетични производни на тестостерон не се превръща в естрогени: етистерон, норетинодрел, даназол. Приемането на тези лекарства по време на бременност може да доведе до вирилизация женски плода.
Неспазването на плацентарна ароматаза
Плацента ароматаза дефицит, причинен от ген мутация CYP19. Обикновено, ембрионите от двата пола ниво андроген секретира от надбъбречната кора и метаболити от него (например, дехидроепиандростерон сулфат) поради повишена физиологичен недостатъчност 3-хидроксистероид дехидрогеназа. Тъй като зародишен дехидроепиандростерон сулфат организъм навлиза в плацентата, където тя се превръща в андростендион и тестостерон, а след това - в естрогени. В недостатъчност плацентна ароматаза ароматизация на андрогени случи. В този случай, както за майката и плода се подложи на вирилизация. Мъж плода с кариотип 46, XY има нормален фенотип, дори когато плацентарна ароматаза дефицит, но майката има признаци на хиперандрогения.
Идиопатична случаи на външните полови органи на междинен тип
В някои случаи, деца с кариотип 46, XX са вулвата междинен вид в отсъствието на всяка друга патология. Преди това, такива случаи се считат за идиопатична, но, както изглежда, вирилизация на гениталиите, причинени от lyuteomoy на бременност (вж. По-горе). Други причини могат да бъдат соматична мутация първи стана мутация или тератогенни ефекти на външни фактори.
 Причините за фетални аномалии. Рискът от фетални малформации
Причините за фетални аномалии. Рискът от фетални малформации Аномалии на SOx гени и синдром TVH Holt-Орам. Фактори фибробласт растежен
Аномалии на SOx гени и синдром TVH Holt-Орам. Фактори фибробласт растежен Количествени патология хромозоми. Качество хромозома аномалия
Количествени патология хромозоми. Качество хромозома аномалия Монозомия х. Associated аномалии монозомия X
Монозомия х. Associated аномалии монозомия X Bookmark половите клетки. определяне хромозомна пол
Bookmark половите клетки. определяне хромозомна пол Вторият мейотичен деление. Значение на мейозата в развитието на зародишните клетки
Вторият мейотичен деление. Значение на мейозата в развитието на зародишните клетки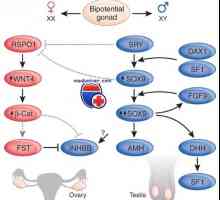 Мутация и ген дублиране dax1, sox9. пол генотип XY несъответствие и kampomelicheskaya дисплазия
Мутация и ген дублиране dax1, sox9. пол генотип XY несъответствие и kampomelicheskaya дисплазия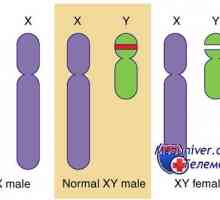 Генетични заболявания на половите жлези. Гените КРЗ, WT1 и синдроми Фрейзър и Денис-dresha
Генетични заболявания на половите жлези. Гените КРЗ, WT1 и синдроми Фрейзър и Денис-dresha Нематода-стоп-половата синдром. Генетика на мъжкото безплодие
Нематода-стоп-половата синдром. Генетика на мъжкото безплодие Две удивителни клетки: яйца и сперма
Две удивителни клетки: яйца и сперма Хромозомни аномалии на плода
Хромозомни аномалии на плода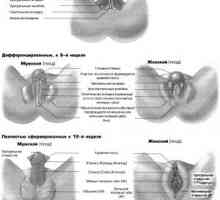 Развитието на половите органи при плода от седмица
Развитието на половите органи при плода от седмица Сперма вашия съпруг определя пола на вашето бебе
Сперма вашия съпруг определя пола на вашето бебе- Вродени нарушения на сексуалното differentsirovkizabolevaniya причинени от хромозомни аномалии.…
- Здраве Енциклопедия, болест, лекарства, лекар, аптека, инфекция, резюмета, пол, гинекология,…
- Пушенето води до загуба на у-хромозома при мъже
 Генетична пол, определяне
Генетична пол, определяне Вярно Хермафродитизмът
Вярно Хермафродитизмът Определяне за подове (шпионин като определяне фактор тестис а)
Определяне за подове (шпионин като определяне фактор тестис а) Кси кариотипни 46 мъже
Кси кариотипни 46 мъже Хромозомни аномалии при децата
Хромозомни аномалии при децата