Алпорт синдром причини и симптоми, диагностика и лечение на синдром на Алпорт
Алпорт синдром (семейство гломерулонефрит)
Съдържание
Заболяването е описан за първи път от британския лекар Артур Алпорт през 1927.
Алпорт синдром е много рядко, но в САЩ, че е отговорен за 3% от случаите на крайна степен на бъбречна недостатъчност при деца и 0,2% от възрастните, и се счита за най-често срещаният тип на семейството нефрит.
Наследствено синдром на Алпорт може да бъде различен:
• X-свързана доминиращ (XLAS): 85%.
• автозомно-рецесивно (АССР): 15%.
• автозомно-доминантно (ADAS): 1%.
Най-често Х-свързана форма на синдрома на Алпорт води до краен стадий на бъбречна недостатъчност при хора. Хематурия обикновено се появява при момчетата със синдром на Алпорт през първите години от живота си. Протеинурия обикновено липсва в детството, но това състояние често се развива при мъже с XLAS и при двата пола с АССР. Загуба на слуха и заболяване на очите, никога не открива при раждането - те се появяват в края на детството или юношеството, много преди развитието на бъбречна недостатъчност.
Причините и механизмът на развитие на синдром на Алпорт
Алпорт синдром е причинена от мутации в гена COL4A4, COL4A3, COL4A5, са отговорни за биосинтеза на колаген. Мутациите в тези гени нарушават нормалната колаген тип IV, който е много важен структурен компонент на основни мембрани в бъбреците, очите и вътрешното ухо.Базалната мембрана - Този тънък филм структури, които поддържат тъкан разделени един от друг. Когато нарушава синтеза на колаген тип IV гломеруларна базална мембранна в бъбреците не са нормално състояние да филтрира токсични продукти от кръвта се влива в протеин в урината (протеинурия) и еритроцитите (хематурия). Аномалии на синтез тип IV колаген води до бъбречна недостатъчност и бъбречна недостатъчност, което е основната причина за смърт в синдром на Алпорт.
клиника
Хематурия - е най-честата и ранна проява на синдром на Алпорт. Микроскопични хематурия се наблюдава при 95% от жените и почти всички мъже. Момчетата хематурия обикновено се открива в първите години от живота. Ако момчето за първите 10 години от живота си не е намерен хематурия, американските експерти препоръчват да се помисли, че е малко вероятно, че наличието на синдром на Алпорт.Протеинурия в детството обикновено липсва, но понякога се развива в момчета с Х-свързан синдром на Алпорт. Протеинурия обикновено прогресира. Значително протеинурия при пациенти от женски пол е необичайно.
Хипертонията е често присъства в пациенти от мъжки пол с XLAS и при пациенти от двата пола с АССР. Честотата и тежестта на хипертония се увеличава с възрастта и развитието на бъбречна недостатъчност.
Сензорна загуба на слуха (загуба на слуха) - е характерна проява на синдром на Алпорт, което се случва доста често, но не винаги. Има цели семейства с синдром на Алпорт, които страдат от тежко бъбречно заболяване, но са с нормален слух. Увреждането на слуха никога не се открива при раждането. Двустранна висока честота загуба на слуха невросензорна обикновено се проявява през първите години от живота си, или в началото на пубертета. На ранен етап на заболяването е решена увреждане на слуха само при аудиометрия.
С напредване, нарушения на слуха се простира до най-ниските честоти, включително и човешка реч. След следва да се очаква появата на загуба на слуха на засягане на бъбреците. Американски учени твърдят, че когато с Х-хромозомата на Алпорт синдром, 50% от мъжете страдат от сензорна загуба на слуха след 25 години, а за 40 години - около 90%.
Преден lenticonus (издут централната част на очната леща напред), наблюдавана при 25% от пациентите с XLAS. Lenticonus не е по рождение, но с течение на годините той води до прогресивна загуба на зрението, което води пациентите до често променят точки. Състоянието не е съпроводено с болка в очите, зачервяване или нарушена цветното зрение.
Ретинопатия - е най-честата проява на синдром на Алпорт от страна на органа на зрението, засяга 85% от мъжете с Х-хромозомата форма на заболяването. Външният вид на ретинопатия обикновено предшества бъбречна недостатъчност.
Задна полиморфна дистрофия на роговицата - рядко състояние, със синдром на Алпорт. Повечето от тях не разполагат с никакви оплаквания. L1649R мутация в ген COL4A5 колаген може да предизвика изтъняване на ретината, който е свързан с X-свързан синдром на Алпорт.
Leiomyomatosis дифузен езофагеален и бронхиална дърво - друго рядко заболяване, което се случва в някои семейства с синдром на Алпорт. Тези симптоми се появяват в края на детството и включват нарушено преглъщане (дисфагия), повръщане, болки в епигастриума и зад гръдната кост, често бронхит, задух, кашлица. Leiomyomatosis потвърдена от КТ или ЯМР.
Автозомно рецесивна форма на синдрома на Алпорт
На АССР представлява едва 10-15% от случаите. Тази форма се среща при деца, чиито родители са носители на една от най-засегнатите гени, комбинацията от които причиняват заболяване при дете. Родителите не са симптоми или имат незначителни симптоми, и децата са много болни - симптомите си приличат XLAS.Автозомна доминантна форма на синдрома на Алпорт
ADAS - това е рядка форма на синдрома, който засяга едно поколение след друг, и мъжете и жените страдат еднакво трудно. Бъбречни прояви и глухота напомнят XLAS, но бъбречна недостатъчност може да се появи по-късно в живота. Клиничните прояви ADAS допълнена тенденция към кървене, makrotrombotsitopeniey, синдром на Epstein, неутрофилна присъствие на включвания в кръвта.Диагностика на синдром на Алпорт
• Лабораторни изследвания. Урината: пациенти със синдром на Алпорт, кръв в урината (хематурия) се появява най-често, и високо съдържание на протеин (протеинурия). Кръвните тестове показват, бъбречна недостатъчност.• тъканна биопсия. Бъбречна тъкан, получена чрез биопсия се изследва чрез електронна микроскопия за присъствие на ултраструктурни отклонения. Кожна биопсия е по-малко агресивни, и американски експерти препоръчват да го правят на първо място.
• Генетичен анализ. В диагнозата синдром на Алпорт, ако все още има съмнения след бъбречна биопсия, генетичен анализ се използва, за да се получи ясен отговор. Определени колаген тип ген мутация IV.
• аудиометрия. Всички деца с фамилна обремененост, предполагащи синдром на Алпорт, трябва да бъдат подложени висока честота аудиометрия да потвърдите сензорна загуба на слуха. Препоръчва се периодично мониториране.
• преглед на очите. Разглеждане от офталмолог е важно за ранно откриване и наблюдение на предната lenticonus и други аномалии.
• бъбречна ултразвук. В по-късните етапи на синдром на Алпорт, бъбречна ултразвук помага да се идентифицират структурни аномалии.
Британски експерти, въз основа на нови данни (2011 г.) на генетични мутации при пациенти с Х-свързан синдром на Алпорт препоръчват тест за генни мутации COL4A5, ако пациентът отговаря на поне два диагностични критерии за Грегъри и COL4A3 и COL4A4 анализ ако COL4A5 мутация не Установихме, или се подозира, автозомно наследяване.
Лечение на синдром на Алпорт
Алпорт синдром е нелечима досега. Проучванията показват, че АСЕ-инхибитори могат да намалят протеинурия и забавят прогресията на бъбречна недостатъчност. По този начин, използването на АСЕ-инхибитори при пациенти с целесъобразно протеинурия, независимо от наличието на хипертония. Същото важи и за ATII рецепторни антагонисти. И двата класа медикаменти изглежда да се намали чрез намаляване на протеинурията intraglomerular налягане. Освен това, инхибиране на ангиотензин II, растежен фактор, отговорен за гломерулна склероза, може теоретично да забави втвърдяване.Някои изследователи предполагат, че такролимус е в състояние да намали протеинурия и стабилизиране на бъбречната функция при пациенти със синдром на Алпорт (проучвания, са малки). Но доклади предполагат, че отговор на пациентите до циклоспорин е много променлива, а понякога и на лекарство може да се утаи интерстициална фиброза.
При бъбречна недостатъчност, стандартни терапии включват еритропоетин за лечение на хронична анемия лекарства за контрол остеодистрофия, корекция на ацидоза и антихипертензивни лекарства за контрол на кръвното налягане. Нанася хемодиализа и перитонеална диализа. Пациенти с Алпорт синдром, бъбречна трансплантация не е противопоказан: Опитът трансплантация в САЩ показа добри резултати.
Генна терапия за различни форми на синдром на Алпорт е опция обещаващ лечение, което е сега активно се проучва западни медицински лаборатории.
Споделяне в социалните мрежи:
сроден
 Синдром на катран в плода. AASE синдром, Holt-Орам плода
Синдром на катран в плода. AASE синдром, Holt-Орам плода Спешна грижа бързо priprogressiruyuschem гломерулонефрит
Спешна грижа бързо priprogressiruyuschem гломерулонефрит Автозомно-доминантно поликистоза на бъбреците при деца. Диагностика и лечение
Автозомно-доминантно поликистоза на бъбреците при деца. Диагностика и лечение Автозомно-рецесивно поликистоза на бъбреците при деца. Диагностика и лечение
Автозомно-рецесивно поликистоза на бъбреците при деца. Диагностика и лечение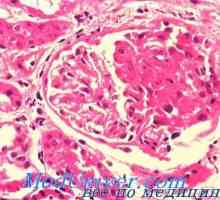 Бързо прогресираща (некротизиращ извънкапилярното) гломерулонефрит. Диагностика и лечение
Бързо прогресираща (некротизиращ извънкапилярното) гломерулонефрит. Диагностика и лечение Клиника на остра бъбречна недостатъчност при деца. диагностика
Клиника на остра бъбречна недостатъчност при деца. диагностика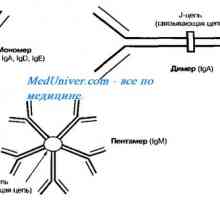 Автозомно рецесивен синдром хиперпродукция на имуноглобулин М (IgM). генна мутация помощ
Автозомно рецесивен синдром хиперпродукция на имуноглобулин М (IgM). генна мутация помощ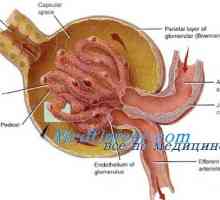 Идиопатична хиперкалциурия при деца. Нефропатия със сърповидно-клетъчна анемия
Идиопатична хиперкалциурия при деца. Нефропатия със сърповидно-клетъчна анемия Остра бъбречна недостатъчност при деца. причини
Остра бъбречна недостатъчност при деца. причини Средно и вроден нефротичен синдром при деца. диагностика
Средно и вроден нефротичен синдром при деца. диагностика Хронична бъбречна недостатъчност при деца. причини
Хронична бъбречна недостатъчност при деца. причини Хроничен тубулоинтерстициален нефрит при деца. Диагностика и лечение
Хроничен тубулоинтерстициален нефрит при деца. Диагностика и лечение- Хематурия и свързаните с болка
- Гломерулонефрит причини и диагностика, лечение и усложнения на гломерулонефрит
- Наследствен нефрит. Етиологията и патогенезата не се разбират. Предполага се, че заболяването е…
- Синдром на системен capillaritis на Goodpasture която засяга предимно белите дробове и бъбреците…
- Здраве Енциклопедия, болест, лекарства, лекар, аптека, инфекция, резюмета, пол, гинекология,…
- Терапия
 Обезболяващо нефропатия: Симптоми, лечение, диагностика
Обезболяващо нефропатия: Симптоми, лечение, диагностика Наследствена нефропатия
Наследствена нефропатия Гломерулонефрит, Лечение, Симптоми, Причини
Гломерулонефрит, Лечение, Симптоми, Причини